Hi,
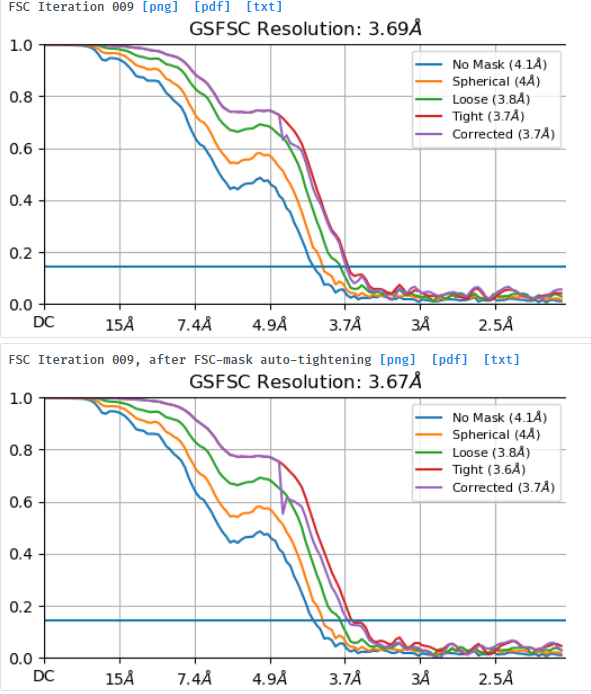
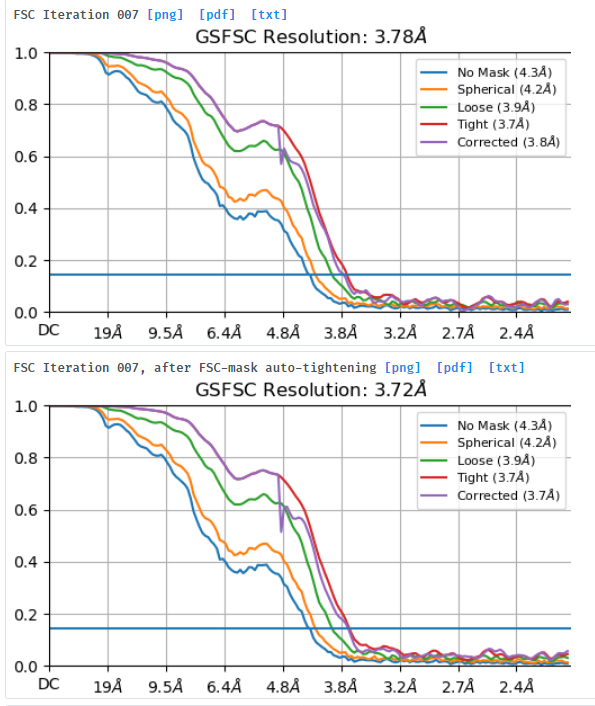
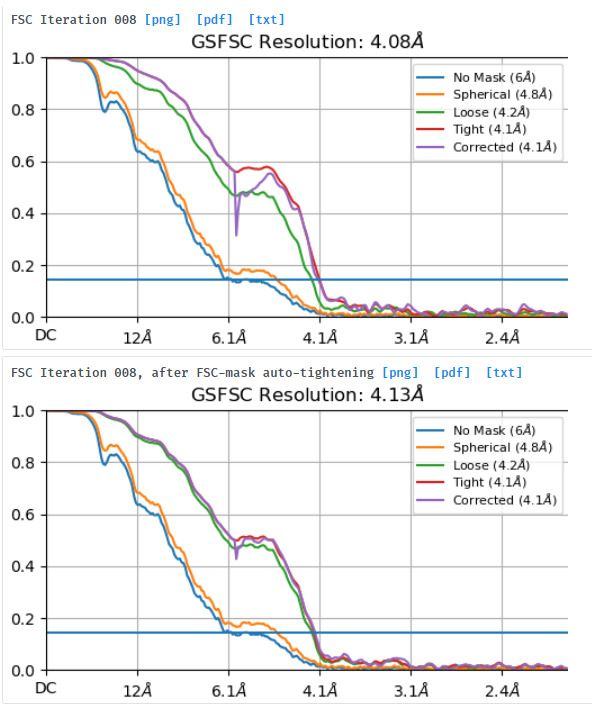
I am processing a dataset that was collected at -1.8um to -3um defocus and am trying to understand why a small box size of 280pix gives better resolution (3.7A resolution) and FSC curves than a box size of 576pix (4.1A resolution). I extracted the same particle stack twice and only varied the extraction box size (pixel size was kept at 1.06apix) before performing NU-refinement on both stacks with the same dynamic masking width. My particle is a glycosylated dimer with two floppy domains and a diameter in the longest dimension of ~220A.
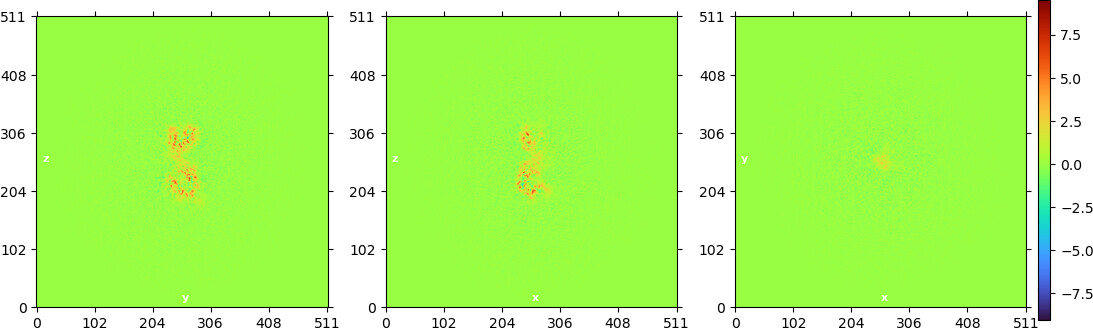
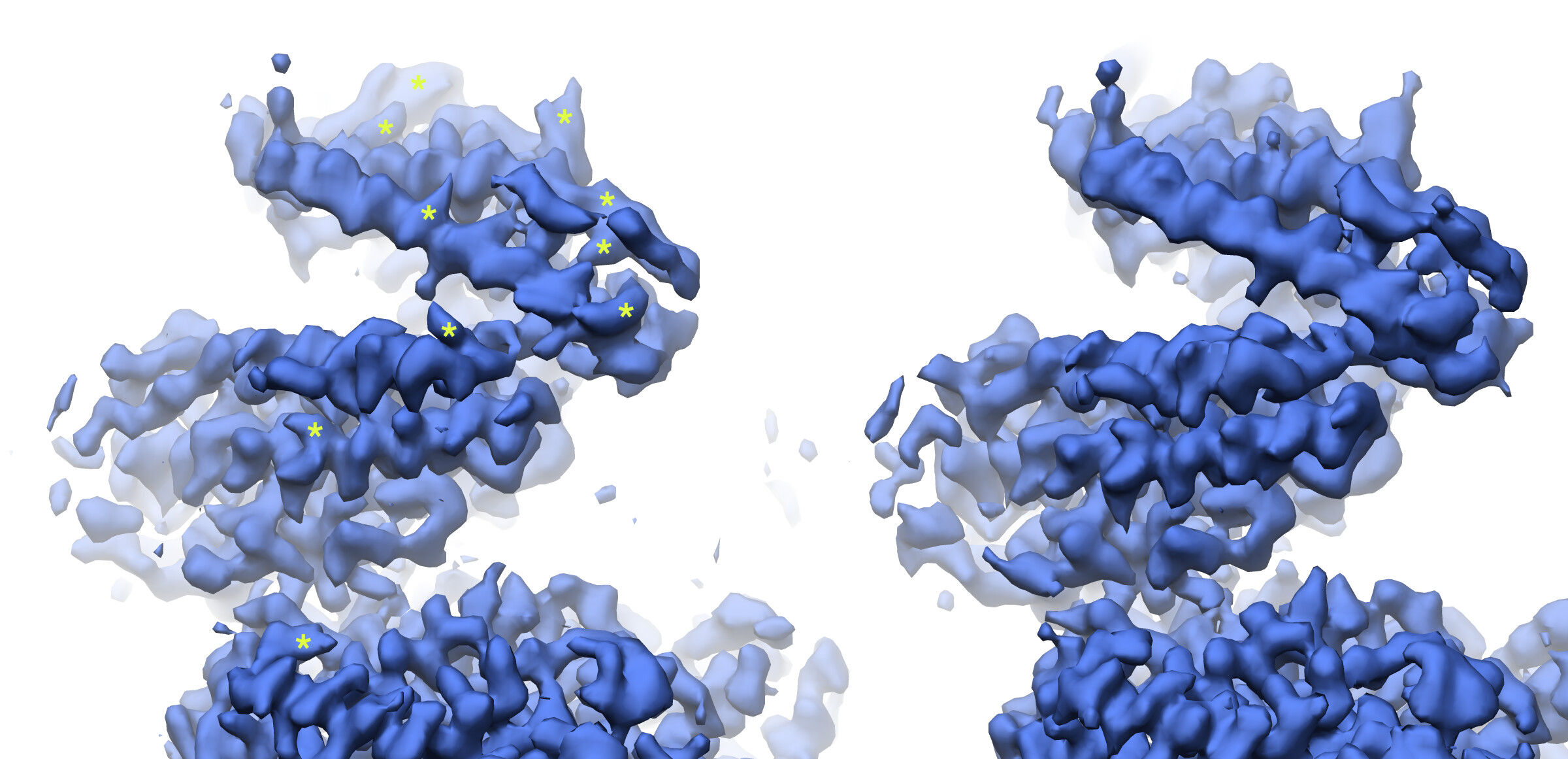
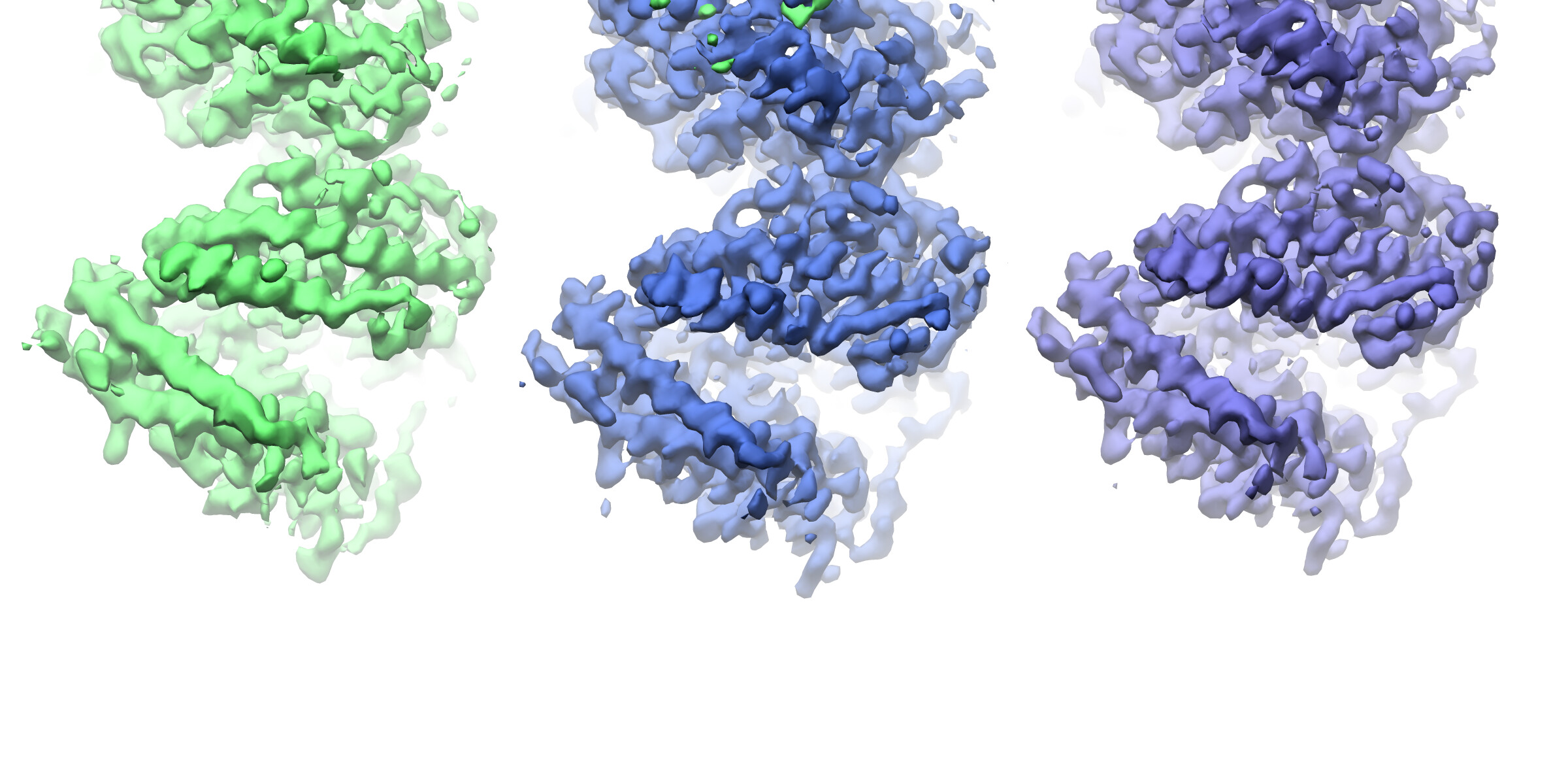
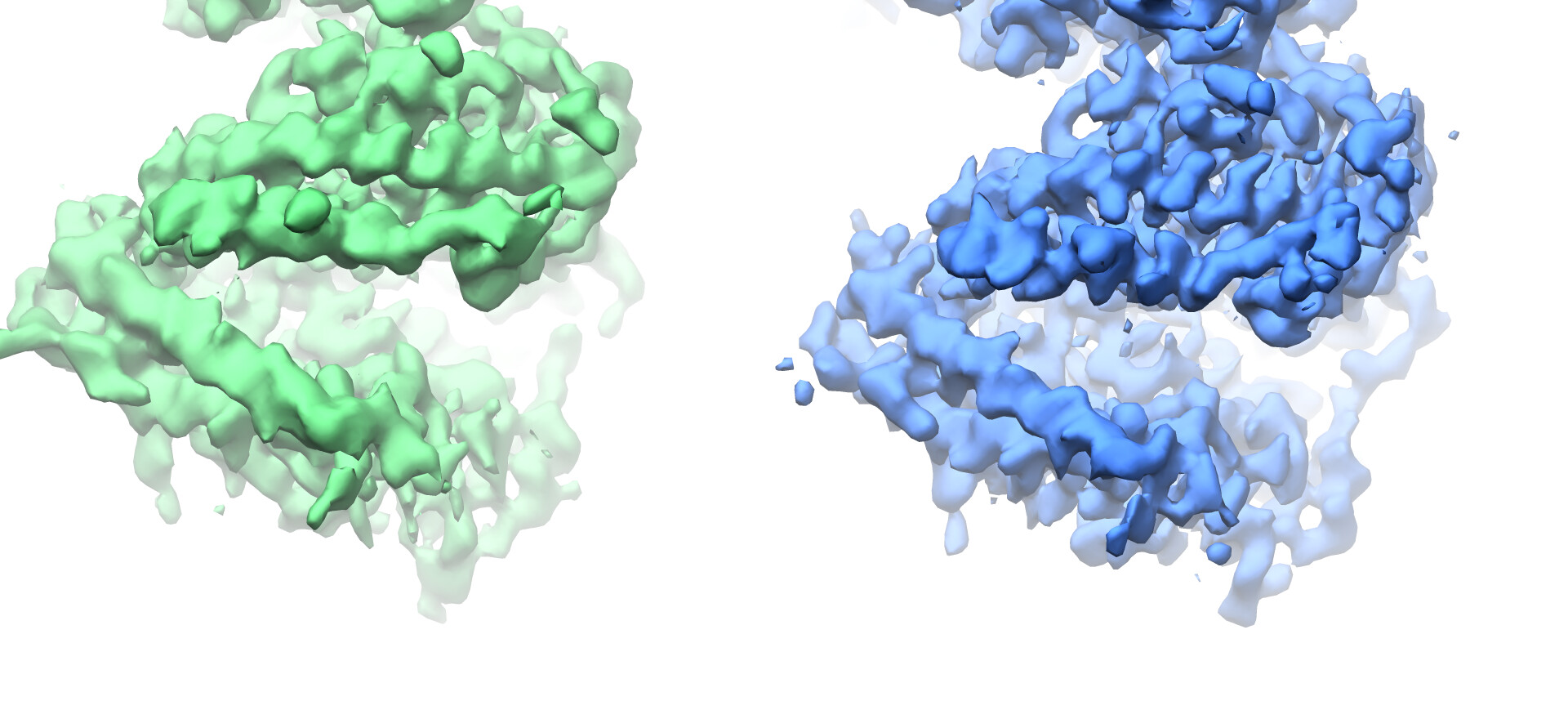
Looking at the maps, I do feel like the improvement in resolution is real:
Green = 576pix box; blue = 280pix box
As mentioned by @olibclarke in this post Cannot align small protein complex particles - CryoSPARC Parameters - cryoSPARC Discuss, the box size to use (in order not to lose information delocalized due to the CTF) is:
B=D+(2XLX(dF/D) (from Rosenthal & Henderson JMB 2003)
I collected data using a Titan Krios thus L ~0.02Å at 300kV.
According to this, should the resolution not increase (or perhaps just stay the same) instead of decrease when I increase the box size?
I thought that a smaller real-space window applied during NU refinement may minimize the effect that an increase in solvent would have on the alignment. Is it possible that the increased noise in a 576pix box in the Fourier domain negatively affects alignment?
What would be the recommended way forward? I feel like the smaller box with better curves should be the obvious choice but want to understand why this happens before moving on.
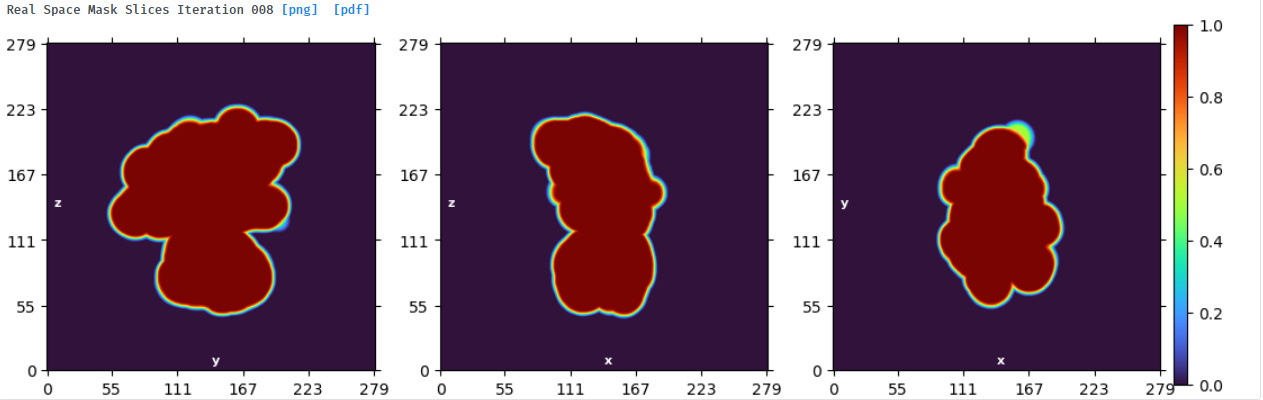
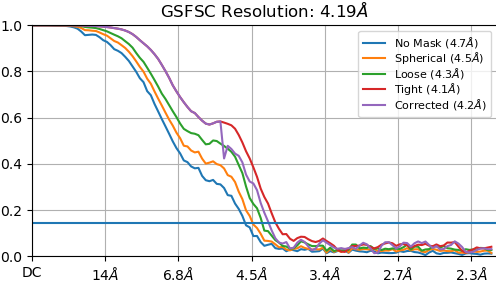
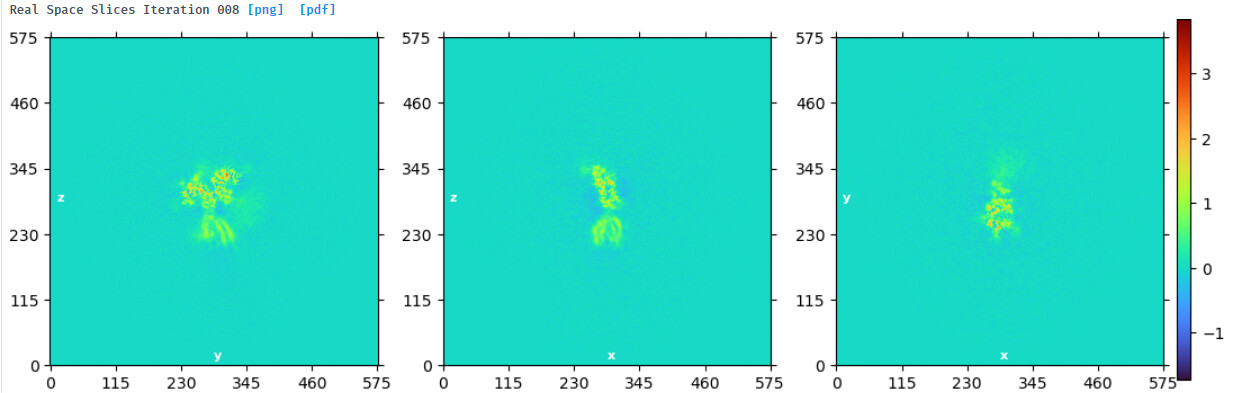
NU refine in 576pix box (real-space window of 0.6-0.75):
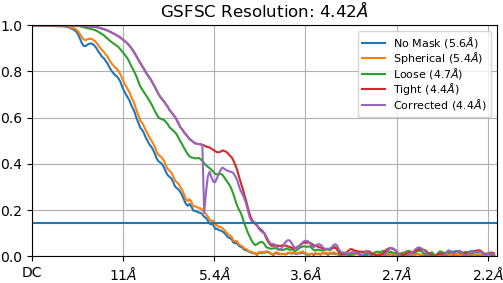
NU refine in 280pix box (real-space window of 0.85-0.99):