I am working on a small membrane protein with no features outside the membrane.
I managed to obtain good 2D classes with the following parameters:
Circular mask diameter (A) = 20% bigger than particle diameter
Force Max over poses/shifts = False
Number of final full iterations = 5
Number of online-EM iterations = 50
Batchsize per class = 150
The transmembrane helices are clearly visible and I can see at least one class (~20.000 particles) with very clear 4-fold symmetry.
In following rounds of 2D classification (as “good” particles are distributed over more classes), classes with 4-fold symmetry become more difficult to identify and, therefore, I was wondering if it would help to increase the “batchsize per class” here? I am inclined to believe that the removal of all bad particles is key for these small membrane proteins.
Currently, I am trying to obtain an initial reconstruction with the following parameters:
Number of Ab-Initio classes = 4
Maximum resolution (A) = 5
Initial resolution (A) = 12
Initial minibatch size = 1500
Final minibatch size = 1500
Class similarity = 0.5
and
Number of Ab-Initio classes = 2
Maximum resolution (A) = 5
Initial resolution (A) = 12
Initial minibatch size = 1500
Final minibatch size = 1500
Class similarity = 0
The main problem here is the lack of noticeable 4-fold symmetry in the ab-initio reconstruction. As some 2D classes are stronger populated than others, is there maybe a way, in cryoSPARC, to select a consistent number of particles per 2D class for the ab-initio reconstruction? Or are there any other ways to solve this problem?
Thanks for your post - this is an interesting case.
The parameters you chose for 2D classificaiton are appropriate for the context.
The batchsize-per-class can help here, and in fact you can turn it up to a very large value (say 1000). This will make the job slower, but it will ensure that the signal from more particles is available at each iteration of classification. You can also play with the “Initial classification uncertainty factor” - a larger value will cause classification to remain uncertain about particle assignments for longer, allowing more exploration to separate otherwise similar particles into more classes.
How sure are you that the protein is actually symmetric? Do you see any other recognizable features in the ab-initio reconstruction? It is important to note that the “resolution” values in ab-initio reconstruction are not resolution estimates and there is no way to objectively measure resolution at the ab-initio stage. Have you tried refining the structures/subsets of particles that you got from ab-initio? This will use a gold-standard split and give some more hints about what is happening in the data.
Otherwise, the parameters you have chosen for ab-initio reconstruction are correct for this context. I’m assuming you’ve already tried with just 1 class and haven’t got something that seemed correct.
For your main question, it is possible (though a little cumbersome right now) to resample particles evenly across views. You can:
Complete 2D classification in a way that resolves the most number of different “views”
Run multiple “Select 2D” jobs from the class 2D output, and in each one, select a different subset of classes, that share a similar view. e.g. select top views, then side views, then front views, etc.
From the output of each Select 2D job, run a “Particle set tools” job. Change the “split batch size” to a number of particles that you wish to take from each separated set of particles (say 10,000) , and turn on “split randomize”. This will randomly select 10k particles from each viewing direction.
Create an abinitio job (or any other that accepts particles) and connect the outputs from all the particle set tools jobs to that job, all connected to the particles input. This job will then operate on a “rebalanced” set of particles.
I have increased the batch-size-per-class to 1000 and
tested the initial-classification-uncertainty-factors: 1, 2, 3, 4 and 5.
“1” helped to remove junk in the first round of 2D classification.
“3” increased the diversity of 2D classes, as expected.
I did not find larger values (“4” and “5”) useful for this particular dataset (the differences between similar classes were small).
2D classes and symmetry:
2D classes containing side views align well and I can see/count the number of transmembrane helices. 2D classes representing other orientations align less well.
Almost all 2D classes show very clear 2-fold symmetry and a few appear to have 4-fold symmetry.
Resampling of particles:
I followed your instructions and did not encounter any problems. So, that was easy to do.
Ab-initio reconstruction:
Unlike my expectations, the single ab-initio reconstruction I generated did not improve with the “rebalanced” set of particles.
If I set the threshold in chimera to a value where all of the micelle is visible, then the overall shape of the reconstruction is as it should be - judging by the visual inspection of the 2D classes - but “internally” I am not seeing the expected number of transmembrane helices or a clear symmetry.
Planned next steps:
I am currently testing NU-refinement with C2 enforced.
Simultaneously, I am also generating 10 ab-initio reconstructions (which usually helps with less complicated proteins) using all particles and the following parameters:
Number of Ab-Initio classes = 10
Maximum resolution (A) = 5
Initial resolution (A) = 12
Initial minibatch size = 1500
Final minibatch size = 1500
Class similarity = 0.1
The micelle, although not very big, is clearly causing some problems here and there is also the possibility of conformational heterogeneity within the dataset. Therefore, is there anything else I could try or are there any other parameters I could tweak when processing?
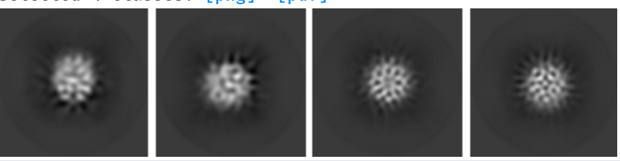
Can you post the 2D class images? I have a small membrane protein with apparent 4 fold and ) 2 fold 2D classes that appear to be a noise artifact . They each have about 20k particles.
Your 2D classes look like noise to me too. How big is your protein?
My protein is over 100 Å in diameter and, therefore, I can exclude any 2D class that looks much smaller.
I have been picking particles from denoised micrographs (collected at high defocus only) and performing 2D classification. I find that this is helpful (for analysis) and I am also picking more orientations.
Hi, I am interesting about the range of the high defocus you collected your micrographs. I’m also working on a small membrane protein. But the helix in the side view is not very clear in the 2D classfication. Would you mind telling the defocus and the molecular weight of your protein?Is the the dimer protein bigger than 100 kD?
 . They each have about 20k particles.
. They each have about 20k particles.