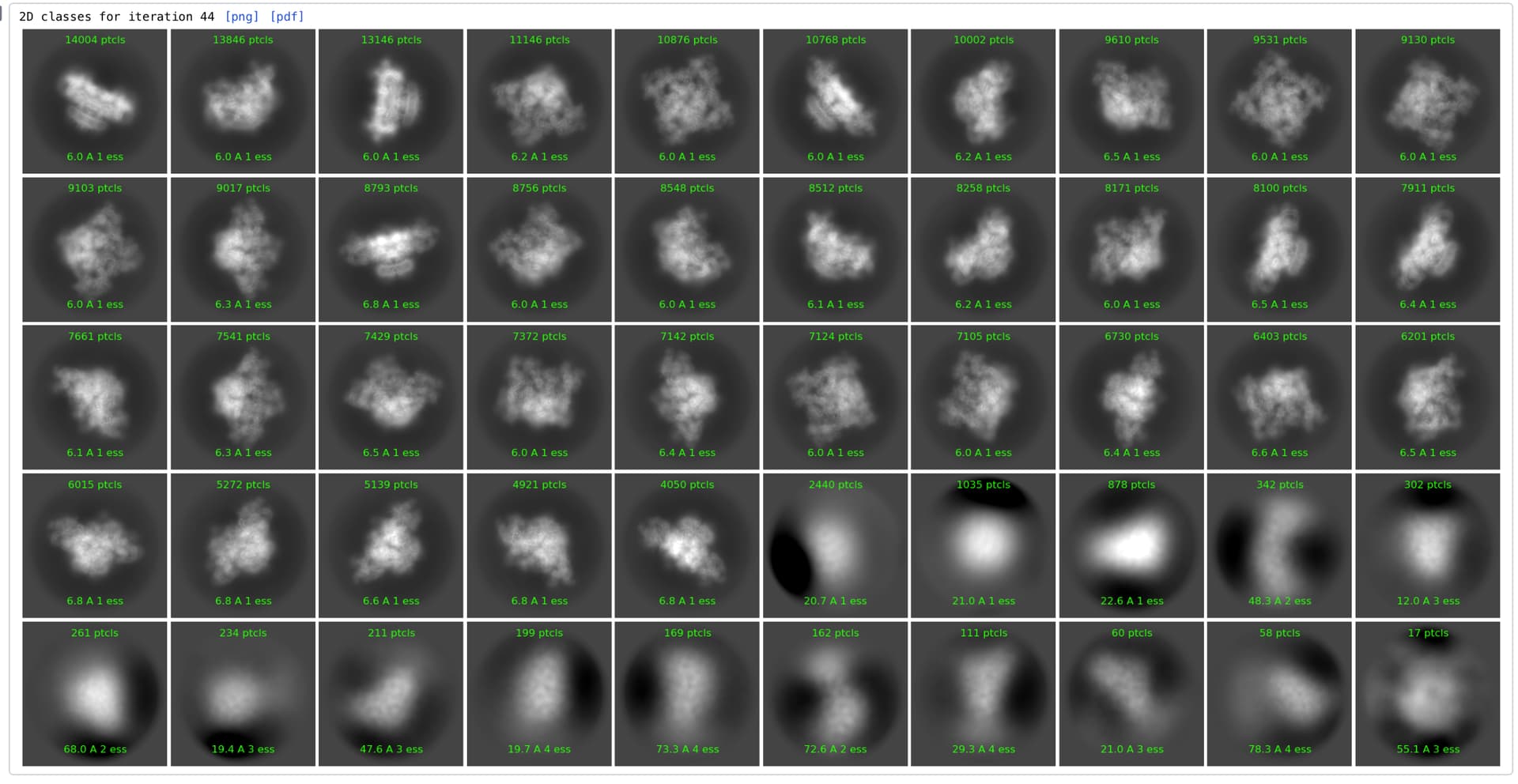
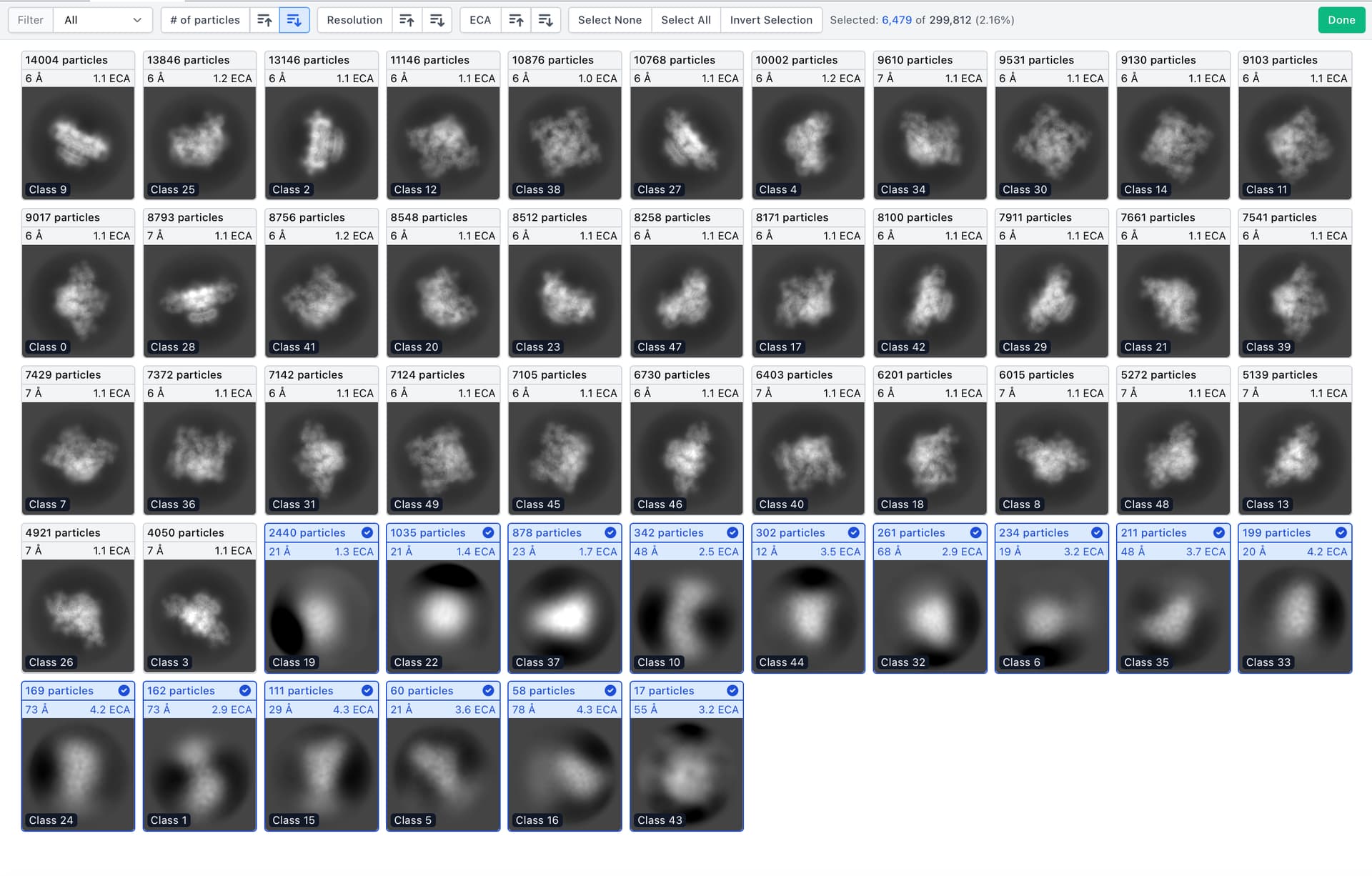
Is there a way to limit the number of particles per 2D class during 2D classification?
We have a protein that has a big and stable part and another big part that moves significantly relevant to the first one and in a subset of the particles falls off. We have cleaned the dataset from junk particles and are trying to disperse the particles among 2D classes but the program over-aligns everything on the first part until the floppy part is averaged out and appears as cleaved off fuzz in 3D that cannot be refined locally or brought back by 3D classification.
During 2D reclassifications we end up with more or less the same number of ‘good’ classes, where the rest remain as classes with zero or few particles even if we double or triple the total number of desired 2D classes (so no gain by increasing the number of classes). We tried playing with the initial uncertainty, masks, align resolution, etc. but cannot really get the particles to not converge. If we select a single class we can break it up in subclasses somewhat more efficiently and we see high resolution features in the floppy domain (so it is not like it is not there) but we would really like to have such dispersal in dataset scale.
Bottomline, is there a way to limit the maximum number of particles per 2D class so that we can force the program to keep things separate and try to align over the entirety of the particles? If not, any other suggestions will be greatly appreciated.
Thanks for posting.
This is definitely an interesting issue.
I think you’ve already tried some of the parameters that would make sense, as well as sub-classification (which is probably the most straightforward way).
Have you also already tried multiclass ab-initio reconstruction of the entire dataset?
With 2D classification, I would suggest trying something like the following settings:
Initial classification uncertainty factor: 3
Number of online-EM iterations: 40
Batchsize per class: 200
Number of iterations to anneal sigma: 35
This will cause the 2D classification job to be quite a bit slower but will more gradually anneal the “certainty” factor so that it takes more iterations before the classes are “locked in”, hopefully giving more time for the separation of very similar classes. You should do this with 200+ classes to ensure there is enough model capacity to find the views you are looking to separate.
Thanks a lot for the suggestion. Just for feedback, after running with the proposed parameters the problem persists and it seems that actually increasing the number of iterations hurts rather than helps : classes that have high-resolution features in the floppy domain appear in the intermaediate iterations and get completely smeared out by the end of the classification. Again in the end there are about 120 ‘empty’ classes (completely empty or with 1-2 particles) out of 200 and the remaining classes are blurred in the flexible region.
Just following up with this - if you can get to 3D, could you try the new 3D Variability algorithm?
Some notes about it here: https://cryosparc.com/docs/tutorials/3d-variability-analysis/
It can generally resolve continuous conformational flexibility quite well and may shed some light into your dataset.
@apunjani , could you suggest parameters, and number of classes, for ab initio of an entire dataset (1 million particles, boxsize 320, pixel size 0.8) ?
Thanks a lot
Jacopo
For very large datasets like this, we usually recommend doing a multi-stage process:
do ab-initio runs with 1, 3, and 6 classes simultaneously, but set the Num particles to use to 100,000 so that the job does not see the entire dataset. Generally you don’t need all 1M particles to find the heterogeneous classes. For the ab-initio reconstruction, default parameters should be okay but if you are working with a very small protein, increase the Maximum resolution and Initial resolution to high resolutions.
from the ab-initio runs, you can select the run that gave the best spread of different conformations (at low resolution) and connect all of the 1M particles and the volumes from ab-initio (say 6-class) to a heterogenous refinement job. This job will process the 1M particles much faster than ab-initio reconstruction but will be able to resolve 6 (or more if you chose) different conformations.
Take the best classes from hetero refinement that all contain the same particle in different conformations (excluding the junk classes) and combine all the particles together as the input of a consensus homogeneous refinement. This will give a single refinement with orientations against a single volume.
Take the refinement output particles and use them to run 3D variability. This will resolve continuous and discrete conformational changes. You can then use 3D variability display to separate clusters, or create intermediate reconstructions along a flexible dimension.
We had a similar issue recently, leading (in retrospect!) to me finding this post - posting my experience here in case it is useful to anyone in the future.
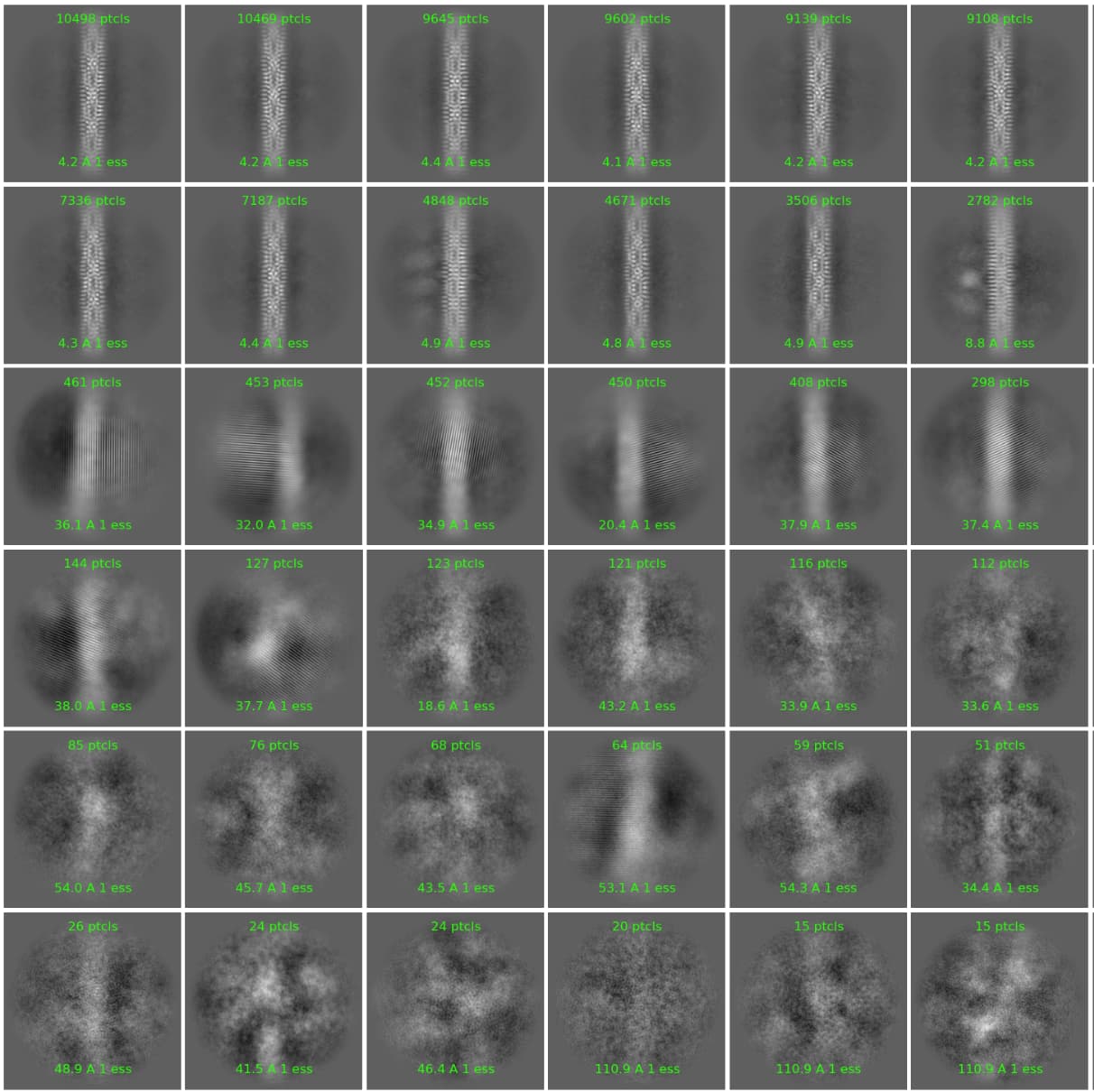
We find that there is frequently a strong “attractor” effect in 2D classification in CryoSPARC, which seems to be related to the sigma annealing process, and which doesn’t seem very sensitive to the initial classification uncertainty factor.
Basically, for a large particle that goes to high resolution on gold grids, we see large junk classes in the first couple of iterations that seem to correspond to gold edges or other junk, but these classes gradually become smaller, and completely disappear by the time sigma has finished annealing - all the particles end up in “good”, high resolution classes.
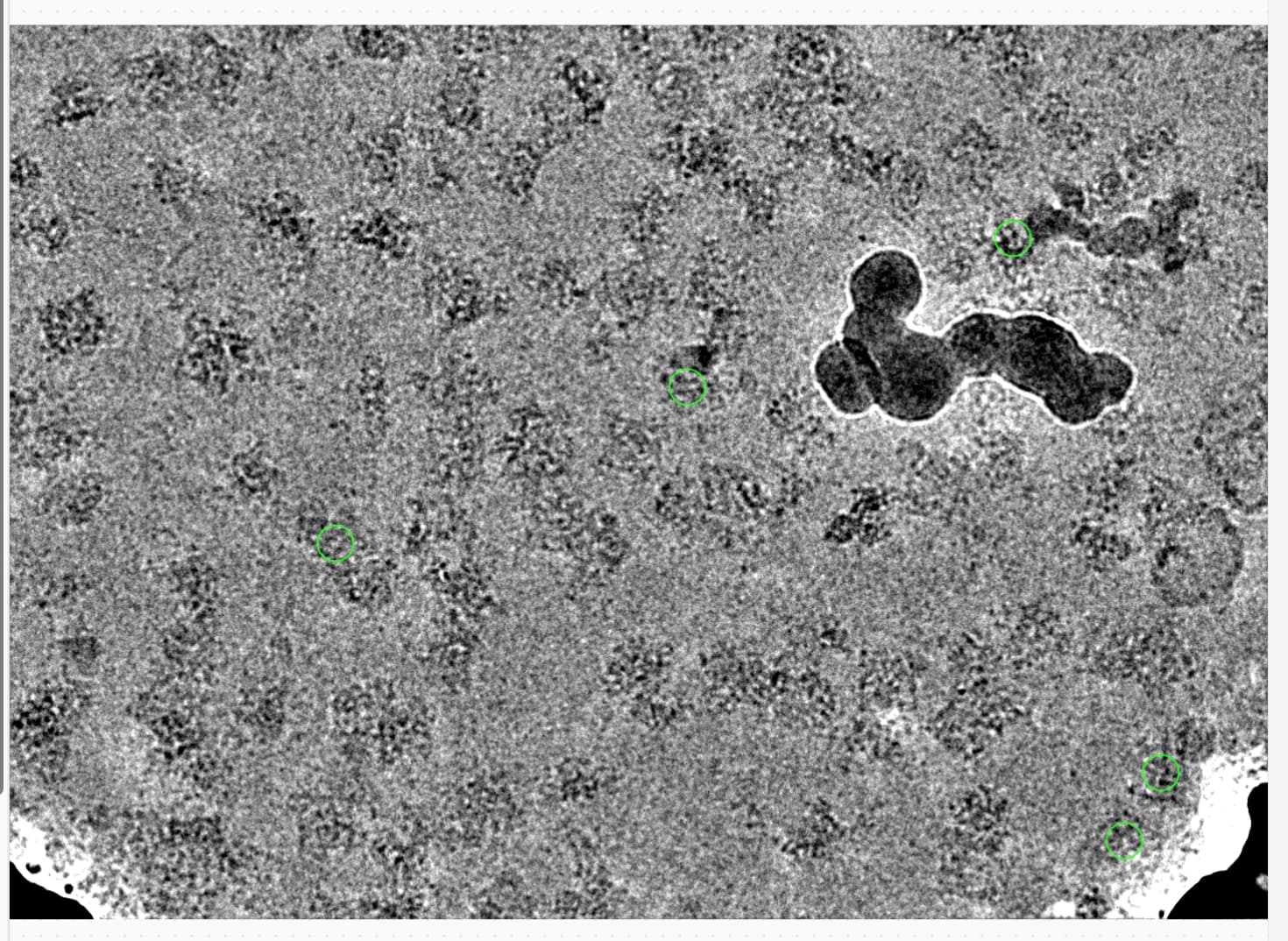
If we switch sigma annealing off completely however, (by setting the iteration to start annealing to higher than the total number of iterations), these classes remain separated, and sure enough when we map them back to the micrographs they correspond to junk! Mostly intact particles but right next to gold edges, or overlapping with surface contaminants.
We have also found the same approach helpful when we have a mixture of particles, some of which are better ordered than others - otherwise the less well ordered ones have a habit of getting subsumed by the better ordered species.
There are now several thousand particles in bad classes. These particles are in fact bad, as we can see looking at particles or micrographs, they are all next to gold or junk:
In this case, another round of 2D with sigma annealing switched off, and recentering switched off, allowed identification of an additional set of bad particles, which were otherwise subsumed in the good classes.
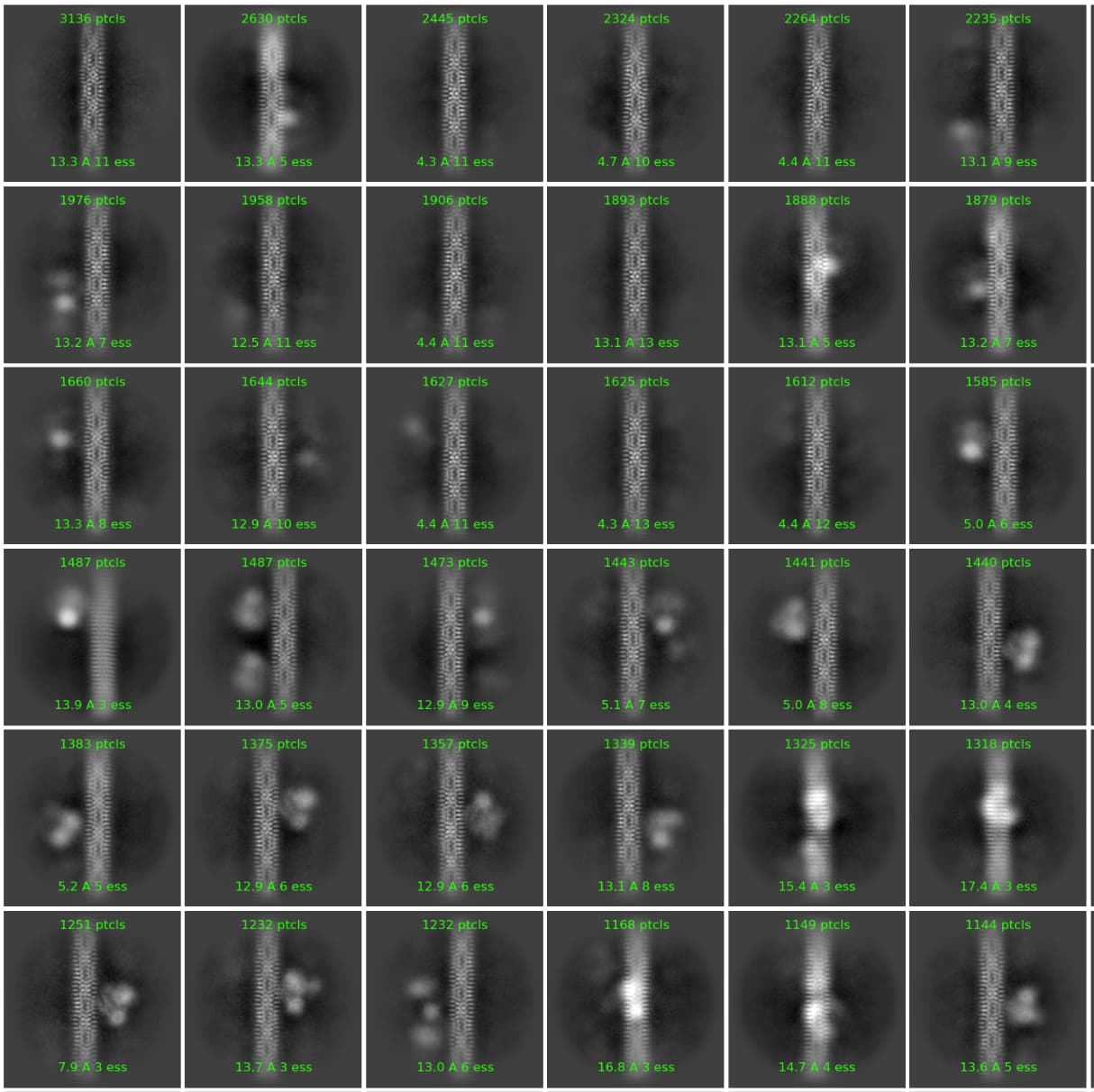
Thanks for posting this investigation, I’m sure this will be quite useful for posterity! We’re increasingly noticing that the definition and estimation of the noise model (sigma) is crucial to the outcome of classification, and it’s interesting to see a case where this has particularly important practical consequences. It will be on our radar for future investigation.
Classification parameters and the input particle set were otherwise identical - it is clear in this case that the default noise model parameters are allowing less occupied or lower resolution classes to be sucked into the majority class, obscuring binding of a protein to the filament.
Hello @olibclarke ,
Amazing finding here. I am able to benefit from your method of turning off sigma annealing off to clean off some bad classes.
I was wondering how are you able to map the particles back to the micrographs? In my experience with CryoSPARC, I was never able to export a particle file that is readable by other software. Or is there a method in CryoSPARC that allow me to map the particles back to the micrographs?
Glad it helped! You should be able to convert the particle.cs file to a star file using the csparc2star.py utility from the pyem package, written by @DanielAsarnow, and you should be able to find further information elsewhere on the forums, hope that helps!