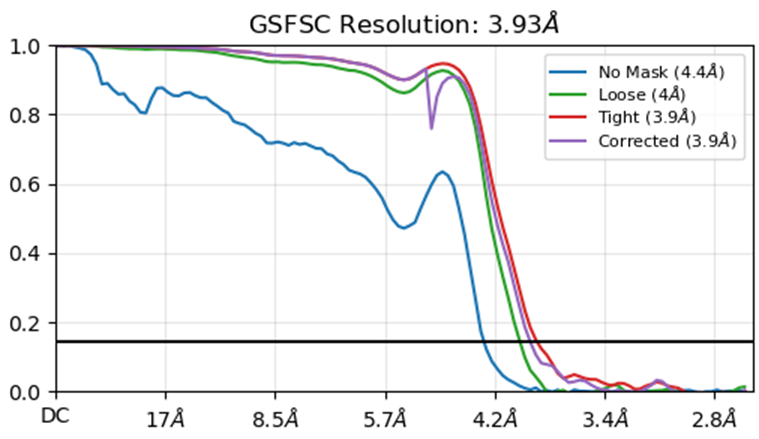
I am working on a beta-barrel protein with the barrel diameter of ~50 Å. It turned out that the particles are very difficult to be aligned. But to my surprise, by squeezing the resolution to ~3 Å using the Ab-initio reconstruction in cryoSPARC, I was able to generate a model with distinguishable beta-strands in the barrel. Though further non-uniform refinement or local refinement always produce much worse results than the ab-initio model. All the beta-strand features were lost. I have trialed with changing the parameters in non-uniform and local refinements, as discussed in the forum. I also tried with applying different masks for the refinement. So far, there is no magic. I also tried to limit the initial lowpass resolution to 6 Å in the local refinement and use gaussian prior of 3°/2Å. This seems helped retain the beta-strands and resulted in a similar final model as the ab-initio. Though the reported resolution and FSC (see below) seem suspicious. Could anyone comment on the results and the settings I used, especially the 6 Å initial lowpass for the local refinement. Is this acceptable? Also, any suggestion on more strategies for dealing this problem is very much appreciated.
Have you tried going directly from ab initio to local refinement? It can be helpful in cases like this. You will need to enable the parameter to redo the GS-split in local refinement.
Thank you @olibclarke for your prompt reply. Yes, thanks to your previous posts on this, I have tried directly go to local refinement from ab initio. The result shown above was the one. My concern is that I have to use an initial lowpass resolution as high as 6 Å to retain the beta-strands. I tried with 8 Å, but all the sheets were lost. I am not sure if the high initial resolution would introduce bias. Would you have a comment on this? Thank you very much.
So long as the initial map is coming from ab initio, and in the initial map you can see features that are clearly higher resolution than 6Å, then I don’t have a big problem with it. Might want to start from a homogeneous reconstruction of the ab initio particles rather than the ab initio directly.
The only issue is that your half sets will not be truly independent, as they were effectively refined together during ab initio. Possibly one could do two rounds of ab initio, one with each half-set, then recombine them for local refinement using cryosparc tools… @rwaldo?
Hmm…you probably could do this with an intermediate step of aligning your two “half maps” to ensure they’re in register (this alignment is handled in normal refinements by the GSFSC split resolution parameter). I’d split the particles into two half sets using Particle Sets Tool, perform the two Ab initio reconstructions, align one volume and particle stack to the other, then set each particle’s alignments3D/split field to 0 or 1, respectively.
I wonder, though, if you’ve tried changing the Initial lowpass resolution parameter in the Homogeneous Refinement job to a higher resolution (lower numerical value). If you’re using the default lowpass of 20 Å, it’s probably just destroying all of your beta strands so there’s nothing to align to. You could try a couple values (5, 7, 10 Å?) and see if that helps?
Yes agreed, if you haven’t tried altering the initial lowpass in homogeneous or NU-refine, I would definitely try that first! Particularly as you have an ab initio volume with high resolution features to start from.
@rwaldo , I have not tried with Homogeneous refinement, but tried Non-uniform refinement with 6 Å initial lowpass, though it did not work. Local refinement worked well with 6 Å and the same particle set, input volume and mask.
@rwaldo Could you please explicit a bit more about this? I thought I understood, but more information would be helpful. Specifically, I can align the volumes/particles using Volume Alignment Tools. Where/how to set the alignments3D/split to 1 or 0?
Yes of course, apologies. You’ll have to use cryosparc-tools to do this. If you haven’t used it before, you can find some guidance on installing and running it here and also here.
Once you have cryosparc-tools working, you can change the value in the fields relatively easily. Here is an example script that should work (using instance-info.json, described in the second link above):
from cryosparc.tools import CryoSPARC
import json
from pathlib import Path
with open(Path('~/instance-info.json').expanduser(), 'r') as f:
instance_info = json.load(f)
cs = CryoSPARC(**instance_info)
assert cs.test_connection()
project_number = "P337"
workspace_number = "W14"
project = cs.find_project(project_number)
abinit_1_uid = "J119"
abinit_1_job = project.find_job(abinit_1_uid)
abinit_1_particles = abinit_1_job.load_output("particles_class_0")
aligned_abinit_2_uid = "J121"
aligned_abinit_2_job = project.find_job(aligned_abinit_2_uid)
aligned_abinit_2_particles = aligned_abinit_2_job.load_output("particles_aligned_0")
abinit_1_particles["alignments3D/split"] = 0
aligned_abinit_2_particles["alignments3D/split"] = 1
combined_particles = abinit_1_particles.append(aligned_abinit_2_particles)
external_job_uid = project.save_external_result(
workspace_uid = workspace_number,
dataset = combined_particles,
type = "particle",
name = "particles",
title = "Combined particles"
)
project.find_external_job(external_job_uid).log(f"Combined particles from {abinit_1_uid} and {aligned_abinit_2_uid} as half-sets.")
In this case,
abinit_1_uid should be the UID (like “J119”) of one of your ab initio jobs
aligned_abinit_2_uid should be the UID of the Align 3D Maps job used to align the other abinit map to the first.
This will produce a new job which has the two particle sets, split correctly, as a single “particles” output:
You should be able to use these particles in a Local Refinement with one of your maps as the input volume. Be sure that Force re-do GS split is turned off!
I’ll be interested to hear how this technique works out for you!
Also, do you think you could share images of the ab initio results and the homogeneous refinement with an initial lowpass filter of 6 Å? I’m curious if I’ll be able to see anything that might indicate to me why the homogeneous refinement is not able to align the particles.
@rwaldo Sorry for the late response, and thank you so much for the invaluable suggestion with all the details, I will give a try with the cryoSPARC tools and let you know.
@rwaldo Sorry for the long delay. I finally managed to have the cryosparc tools installed and tested running with the script. Though there are some errors reported.
Connection succeeded to CryoSPARC command_core at http://rohpc02:39002
Connection succeeded to CryoSPARC command_vis at http://rohpc02:39003
Connection succeeded to CryoSPARC command_rtp at http://rohpc02:39005
/usr/local/biotools/python/3.11.5/lib/python3.11/contextlib.py:137: UserWarning: *** CommandClient: (http://rohpc02:39003/external/projects/P17/jobs/J210/outputs/particles/dataset) URL Error [Errno 104] Connection reset by peer, attempt 1 of 3. Retrying in 30 seconds
return next(self.gen)
/usr/local/biotools/python/3.11.5/lib/python3.11/contextlib.py:137: UserWarning: *** CommandClient: (http://rohpc02:39003/external/projects/P17/jobs/J210/outputs/particles/dataset) URL Error [Errno 104] Connection reset by peer, attempt 2 of 3. Retrying in 30 seconds
return next(self.gen)
Traceback (most recent call last):
File “/rodata/cryoem/m060907/GM146-1/CS-gm146-1/combineParticles.py”, line 29, in
external_job_uid = project.save_external_result(
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “/home/m060907/.local/lib/python3.11/site-packages/cryosparc/project.py”, line 278, in save_external_result
return self.cs.save_external_result(
^^^^^^^^^^^^^^^^^^^^^^^^^^^^^
File “/home/m060907/.local/lib/python3.11/site-packages/cryosparc/tools.py”, line 584, in save_external_result
job.save_output(output, dataset)
File “/home/m060907/.local/lib/python3.11/site-packages/cryosparc/job.py”, line 1520, in save_output
with make_request(self.cs.vis, url=url, data=dataset.stream()) as res:
File “/usr/local/biotools/python/3.11.5/lib/python3.11/contextlib.py”, line 137, in enter
return next(self.gen)
^^^^^^^^^^^^^^
File “/home/m060907/.local/lib/python3.11/site-packages/cryosparc/command.py”, line 226, in make_request
raise CommandError(error_reason, url=url, code=code, data=resdata)
cryosparc.errors.CommandError: *** (http://rohpc02:39003/external/projects/P17/jobs/J210/outputs/particles/dataset, code 500) URL Error [Errno 104] Connection reset by peer
Could you help suggest a solution to the error? Many thanks again!
Is the version of cryosparc-tools the same as the version of CryoSPARC you’re running? E.g., the first two numbers in the top-left of your CryoSPARC home page:
should be the same as the result of pip freeze | grep cryosparc
(note that CryoSPARC is 4.5.3, while tools is 4.5.0. That’s okay, it’s just the first two numbers that need to match).
If they don’t match, try running pip install --upgrade cryosparc-tools~={version}, replacing {version} with your CryoSPARC version number with the last number replaced with a zero. For example, in this case:
I’d run pip install --upgrade cryosparc-tools~=4.4.0.
I realized that it is possibly the ‘save_external_result’ that cannot be executed.
external_job_uid = project.save_external_result(
workspace_uid = workspace_number,
dataset = combined_particles,
type = “particle”,
name = “particles”,
title = “Combined particles”
)
I may be wrong, but is there any possibility that it is related to the internet or firewall settings, or it is more likely a problem with Python settings? Any clue is very much appreciated!
@rwaldo I was able to install the cryosparc-tools on our another workstation and have it run with the script you previously shared (thank you so much for this!). Though I got a different error than the previous one when running on the HPC. Please see the details below. Could you please suggest how I can fix the error? Thank you for any comments/suggestions.
[mxxxxx@cxxxxx CS-test]$ python reCenter.py
Connection succeeded to CryoSPARC command_core at http://cxxxxx:61002
Connection succeeded to CryoSPARC command_vis at http://cxxxxx:61003
Connection succeeded to CryoSPARC command_rtp at http://cxxxxx:61005
/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/contextlib.py:113: UserWarning: *** CommandClient: (http://cxxxxx:61003/external/projects/P13/jobs/J160/outputs/recentered_particles/dataset) HTTP Error 422 UNPROCESSABLE ENTITY; please check cryosparcm log command_vis for additional information.
Response from server: b"Invalid dataset; missing the following required fields: {(‘location/micrograph_psize_A’, ‘<f4’)}"
return next(self.gen)
Traceback (most recent call last):
File “reCenter.py”, line 93, in
project.save_external_result(
File “/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/site-packages/cryosparc/project.py”, line 278, in save_external_result
return self.cs.save_external_result(
File “/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/site-packages/cryosparc/tools.py”, line 584, in save_external_result
job.save_output(output, dataset)
File “/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/site-packages/cryosparc/job.py”, line 1520, in save_output
with make_request(self.cs.vis, url=url, data=dataset.stream()) as res:
File “/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/contextlib.py”, line 113, in enter
return next(self.gen)
File “/home/mxxxxx/miniconda3/envs/Geeks/lib/python3.8/site-packages/cryosparc/command.py”, line 226, in make_request
raise CommandError(error_reason, url=url, code=code, data=resdata)
cryosparc.errors.CommandError: *** (http://cxxxxx:61003/external/projects/P13/jobs/J160/outputs/recentered_particles/dataset, code 422) HTTP Error 422 UNPROCESSABLE ENTITY; please check cryosparcm log command_vis for additional information.
Response from server: b"Invalid dataset; missing the following required fields: {(‘location/micrograph_psize_A’, ‘<f4’)}"
log of command_vis:
2024-08-06 13:57:01,551 upload_external_job_output INFO | Received external job output P13.J160.recentered_particles
2024-08-06 13:57:01,681 upload_external_job_output ERROR | Invalid dataset; missing the following required fields: {(‘location/micrograph_psize_A’, ‘<f4’)}
3 error ERROR | Can’t connect to (‘0.0.0.0’, 61003)
2024-08-06 12:20:34 info INFO | Starting gunicorn 20.1.0
2024-08-06 12:20:34 error ERROR | Connection in use: (‘0.0.0.0’, 61003)
2024-08-06 12:20:34 error ERROR | Retrying in 1 second.
2024-08-06 12:20:35 error ERROR | Connection in use: (‘0.0.0.0’, 61003)
2024-08-06 12:20:35 error ERROR | Retrying in 1 second.
2024-08-06 12:20:36 error ERROR | Connection in use: (‘0.0.0.0’, 61003)
2024-08-06 12:20:36 error ERROR | Retrying in 1 second.
2024-08-06 12:20:37 error ERROR | Connection in use: (‘0.0.0.0’, 61003)
2024-08-06 12:20:37 error ERROR | Retrying in 1 second.
2024-08-06 12:20:38 error ERROR | Connection in use: (‘0.0.0.0’, 61003)
2024-08-06 12:20:38 error ERROR | Retrying in 1 second.
2024-08-06 12:20:39 error ERROR | Can’t connect to (‘0.0.0.0’, 61003)
Some more update. The error persists when running different scripts from the cryosparc-tools examples and the above script. Although the example script #5 (generate High-Res 2D classes) worked on both the workstation and HPC, without any errors.
More updates. I was able to figure out the errors are not related to Python or internet. Instead, by modifying the ‘save_external_result’ portion of the script, it was run to the end without errors. I inserted
slots = [‘blob’]
to the
project.save_external_result( )
to give
external_job_uid = project.save_external_result(
workspace_uid = workspace_number,
dataset = combined_particles,
type = “particle”,
name = “particles”,
slots = [‘blob’],
title = “Combined particles”
)
Though I severely doubt this is the correct parameter to use. @rwaldo Could you or anyone else please suggest what changes should I make to generate the correct particle stack? Appreciated!
Hi @rwaldo , thank you for your response. I still got errors with the generated particle stack when running local refinement. Specifically, how can I keep the ‘alignment3D’ from the Ab initio jobs?
Traceback (most recent call last):
File “cryosparc_master/cryosparc_compute/run.py”, line 73, in cryosparc_master.cryosparc_compute.run.main
File “/biotools8/biotools/cryosparc/cryosparc_worker/cryosparc_compute/jobs/runcommon.py”, line 1213, in check_default_inputs
assert False, 'Non-optional inputs from the following input groups and their slots are not connected: ’ + missing_inputs + ‘. Please connect all required inputs.’
AssertionError: Non-optional inputs from the following input groups and their slots are not connected: particles.ctf, particles.alignments3D. Please connect all required inputs.