Hello,
I require assistance with a data processing issue that I am currently facing.
We had a dataset with about 11,000 movies and finally I could get a map of it with the resolution of 4.1A (about 40,000 particles). We added another dataset to that included 7000 movies, now the number of particles are about 100,000. However, the resolution is only 3.9A. How can I improve this resolution? I usually do 2D classification, 3D classification, Homo refinement, and Non-Uni refinement. I try to get rid of the bad particles as much as I can in 2D and 3D classification steps. These datasets are related to a membrane protein which is 150 KD in size.
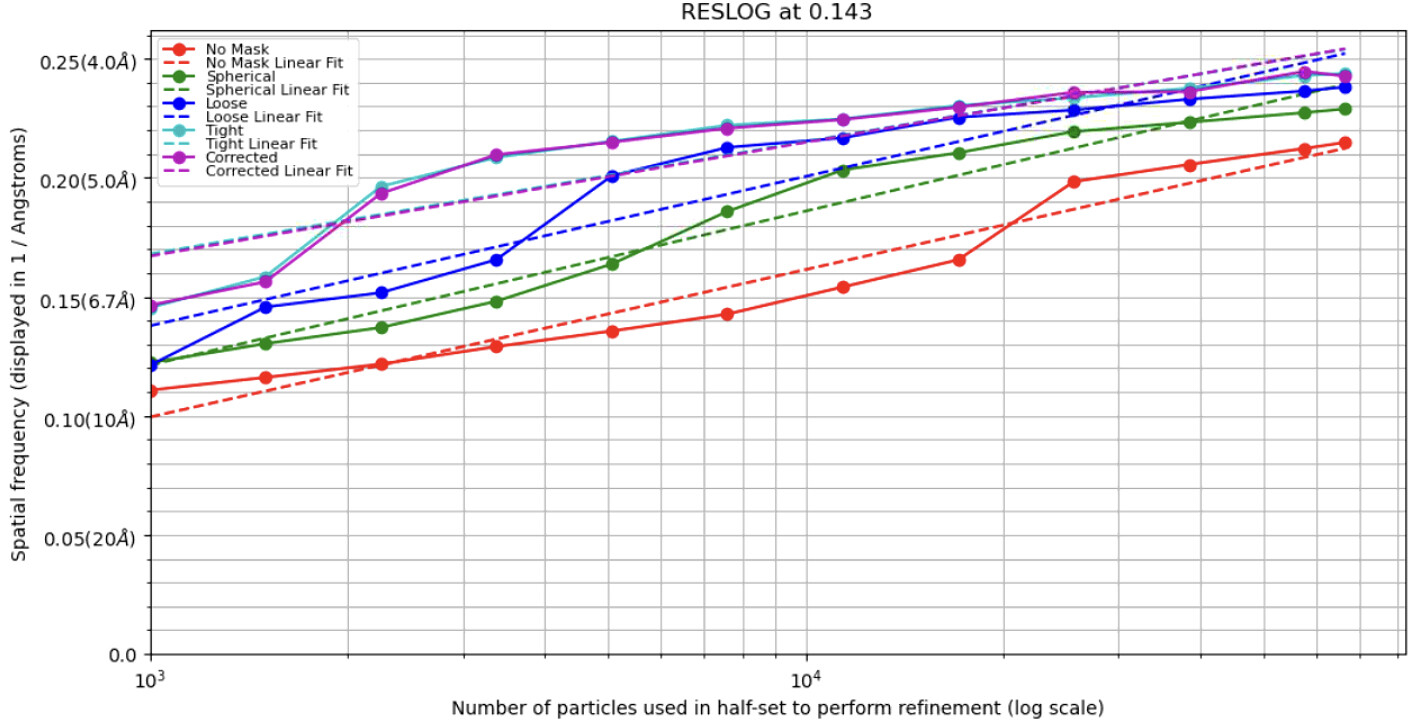
There are so many reasons resolution could be limited. The most obvious is that signal does not increase proportionally to the number of observations (iirc signal = sqrt(# observations), hopefully someone else better steeped in the underlying theory will chime in). So adding n more particles does not mean you have n more signal and correspondingly more resolution. Run a reslog analysis to see what the expected resolution should be of 100,000 particles vs 40,000, and decide if they make sense. If the relationship is linear, you just need more particles. If it’s a plateau, something else is going on that’s limiting resolution.
It’s also possible you have variability or compositional heterogeneity that is limiting your resolution - the place to start there would be a 3D variability job, and simple volume series display, to get an idea about the magnitude of any flexibility or heterogeneity. You may need to sort those to get increased resolution.
3 Likes
@Freza065 40K particles from 11K movies is on the lower side. How are you picking ? (Topaz of CrYOLO can increase accuracy, centering, etc - in many cases). I did not see heterogeneous refinement, are you starting with over 100k particles and using multiclass Ab-initio. How many 3D classes are you using and are they similar in resolution & number of particles ?
Just talking about resolution ? I don’t think upsampling .eer is worth it. Reference based motion correction. 150 KDa could be on the small side for mask + particle subtraction => local refinement. Extraction box size could be optimized. Symmetry expansion if not C1. Manually Curate Exposures => harsh CTF cut off at 3.5A.
Nice video here https://www.youtube.com/watch?v=9GszV5W4riI
I have encountered a similar problem with my protein complex (~200 kDa). Resolution typically peaks around 3.5-4 A. Consider what the typical resolution is reported with this protein. Are there a lot of flexible loops that can make particle alignment difficult? Some things that helped with my processing:
-Use template picking using a good volume from your current processing as a template (use “Create Template” to get started). Clean it up via 2D class. Then pool everything and remove duplicates. Doing this almost doubled my number of particles.
-Some of the biggest improvements I’ve had have come through NU refinement by playing with the parameters, specifically the mask threshold and dilation. Takes some time fine tuning, but well worth it.
-Reference based motion correction actually worked wonders for me for one dataset. I wasn’t expecting much, but it improved my resolution by 0.2-0.3 A.
-It’s always worth trying different box sizes. My samples are all pretty similar sizes, yet they all required different box sizes.
-Agree with @Mark-A-Nakasone that masking and local refinement might not work due to size. Didn’t work for me, but it’s still worth a shot. Maybe aligning onto a more stable/large portion of the protein?
Lastly, and this may seem obvious, but consider the question you’re asking. Do you need a sub 3 A map? Maybe a 4 A map can be just as informative (again, depending on the question(s) you’re asking).
1 Like
Thank you for your comment! I did reblog and the it looks like this:
When I run ab-initio with a couple of classes and similarity factor of 0 between classes, it gives me two classes of my protein. I have checked the maps on Chimera and they look to overlap in all the sections. I am not sure if that is why the resolution doesn’t improve.
Thank you for your comment! Yes, I have started about 400K particles and did ab-initio with different classes. I have tried with different classes like 1, 3, 8.
I also attached my reslog here.
Thank you for your suggestions! There are other resolutions like 3.1A from this protein.
I have repeated my particle picking with different templates and models, finally, I get the same results. With mask threshold and dilation for NU do you suggest to use?
My box size now is 256 and my protein size is about 140A, Do I need to change the box size, like downscaling it?
I have done a local refinement on Phenix and it showed that some parts of the map are 3.5A, but the worse also was 12A.
I have also tried to mask out the detergent belt to see if it helps, but it didn’t really help.
My only suggestion for NU ref mask threshold and dilation would be to try different values and see what works. My protein is heavily glycosylated; there’s a separate thread detailing how fine tuning mask parameters are crucial for these types of proteins and that is what I found as well. I don’t have any experience with proteins in a detergent belt, but it could be the same case. One common denominator for me was to never use a tight mask as this eventually led to losing densities for smaller (15-20 kDa) domains.
It’s definitely worth re-extracting with different box sizes and binning to see what works. I’ve read that starting with a smaller box size and slowly increasing helps. But again, it’s worth trying different sizes/binning to see what works best for your sample.
1 Like