I am working with a 110 kDa monomeric, non-symmetric protein carrying four N-glycans. I get good 2D classes and reasonable ab initio models, but when I refine (NU refinement) I get shell-like artifacts presumably originating from the N-glycans, protruding out almost like funnels and then covering part of the protein. Do you have any suggestions regarding how to mask out the glycans in order to increase the details of the protein core?
Hi @emil ,
I have a similar protein to yours (~140kDa monomeric, non-symmetric, ~10 N-glycans). My 2D classes showed very clear secondary structure details and fuzzy density on the surface due to glycans. Getting a decent 3D structure has proven to be very difficult for a small protein with such high glycosylation. I had to be sure that all junk was removed and that all particles were monomeric - 3D initial model generation otherwise just gave a smear. I still have some internal heterogeneity (hinging and glycan motion) but have now managed to get a moderate resolution 3D structure with better resolution at the protein core (really looks like ~4/5A).
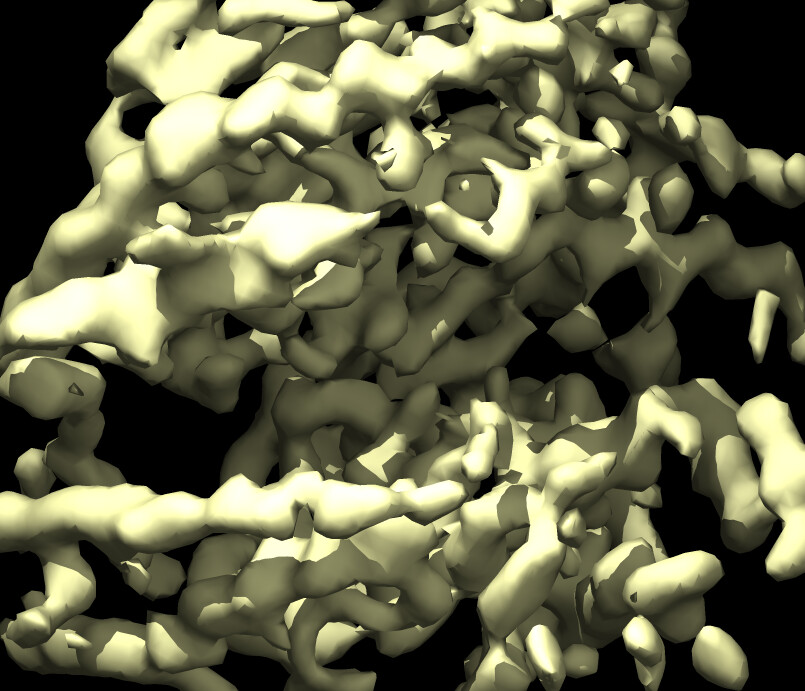
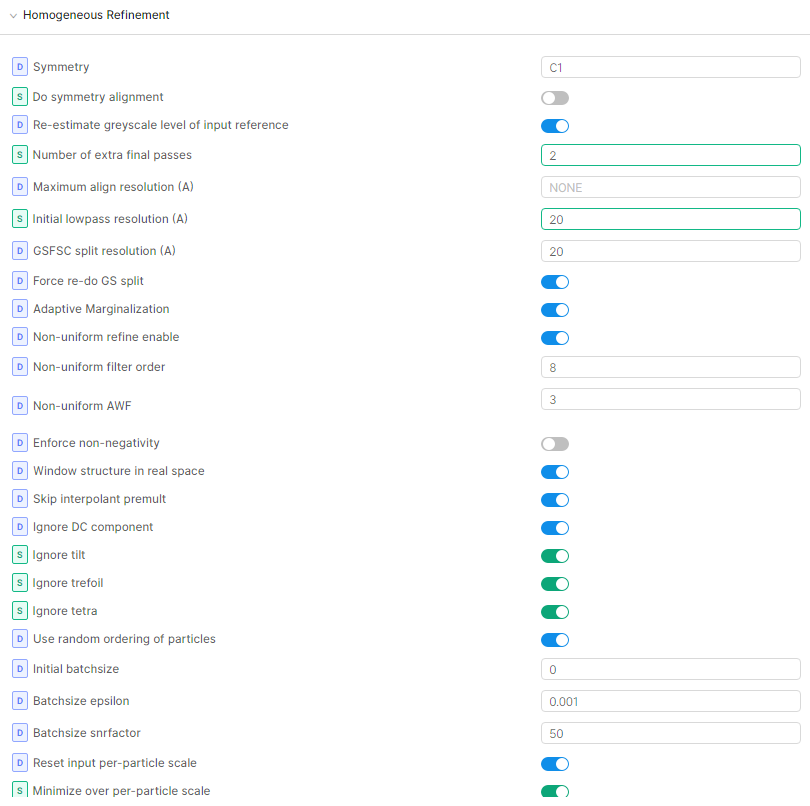
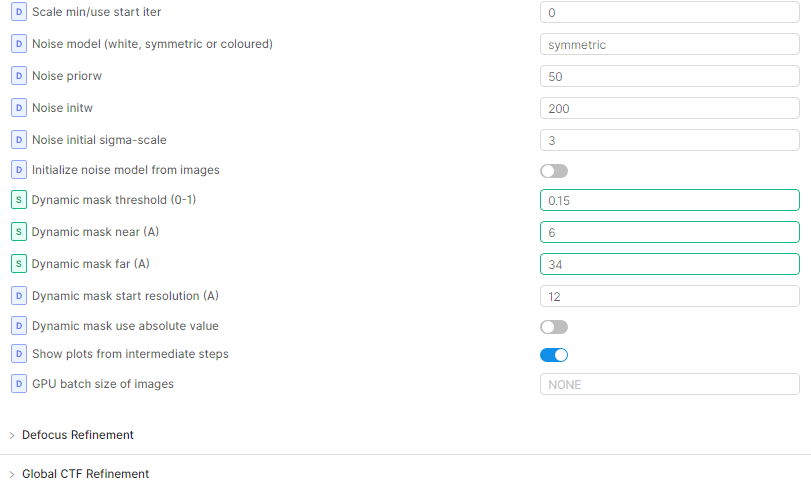
Glycans are very flexible and they may influence the alignment of your particles in 3D. In my case, the glycan motion occurred along with protein motion so that there was a shell of density on the surface, although resolution reported with FSC curves wasn’t bad. What worked for me was firstly to extend the dynamic mask radius. I have branched complex-type N-glycans and the default of 6A near and 14A far was too tight and parts of the density was cut away (that’s what I think anyway Local B-factor sharpening - Post Processing - cryoSPARC Discuss). Extending the radius to 12A near and 20A far seems better and gave glycan stumps on the surface of lower resolution than the protein core.
To improve the resolution beyond what I get with a consensus refine of all the good 2D classes’ particles, I did ab initio with 4 classes followed by heterogeneous refinement. This gave an improvement which was then even better with non-uniform refine. I only see good glycan density when the resolution overall is rather low. With sharpening they are basically lost.
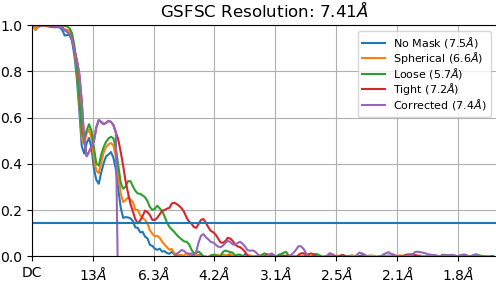
Here is a pic after NU refine at low and high thresholds showing glycans before sharpening and core density after sharpening.
Hoping that this may give you some tips - although I am still in need of some myself. I thought that as long as I get rid of as much protein heterogeneity as possible, glycan heterogeneity would not influence the 3D alignment as much. Perhaps we can get some information about the glycans at the end by using per-pixel B-factor sharpening or something like that? I am trying to use 3D variability analysis to further ‘purify’ my current classes so that I can reconstruct the most homogenous set of particle conformations but not sure how to focus the analysis on protein variability and not glycan variability (Masking glycans for 3D variability analysis - 3D Variability Analysis - cryoSPARC Discuss) Maybe something like that could help you as well?
I am not an experienced structural biologist (this is the first cryo EM dataset I have ever worked with), but I would say that the protein core is NOT resolved at 4.3 Å. I know (from a crystal structure) what 80% of the structure should look like, but I can’t seem to find those details in the map. In fact, the 2D classes are actually revealing more details…
Thanks a lot for a detailed walkthrough of your processing steps. It is definitely of great value to me at this point!
I have not tried extending the mask radius, but it makes complete sense! I will try it out.
I just tried decreasing the maximum/initial resolution of the ab initio job to 5/7 Å and that helped a lot with increasing the resolution of the protein core in subsequent steps. Hopefullly a combination of the mask dilation and the new ab initio models will improve the refined structure.
I had roughly 400k particles before splitting into 4 classes with ab initio and hetero refine. The best class had around 130k and these were used for nu refine. But choosing 4 classes was a guess and I may still need further subclassification. I am hoping to get insight into the degree of heterogeneity remaining within the current classes by doing 3dva
I tried running NU refine with expanded mask, but unfortunately it did not seem to help.
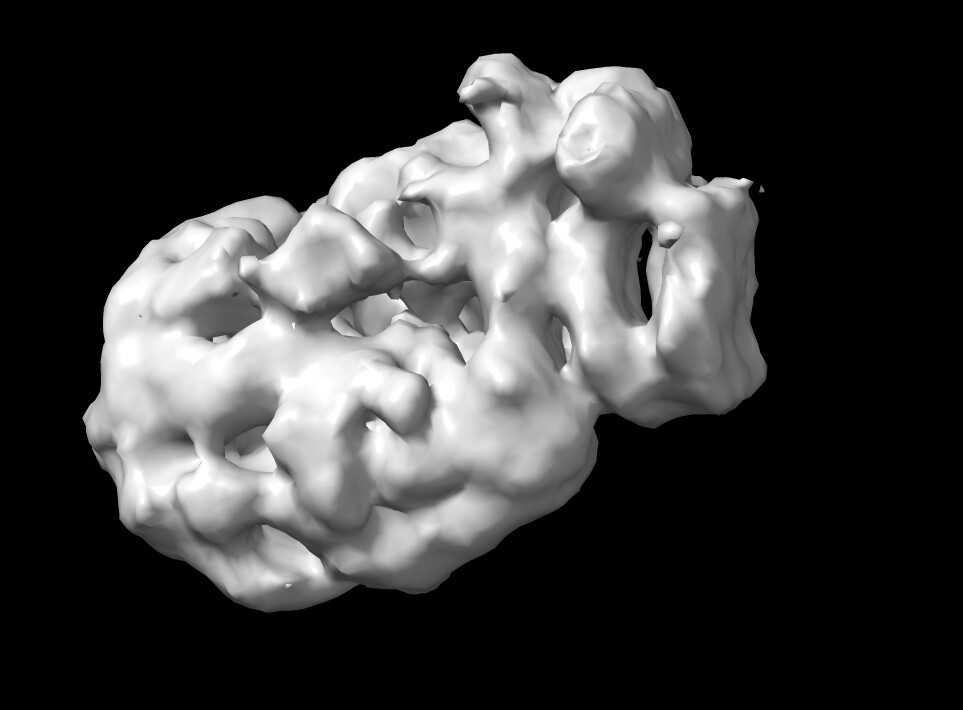
This is what the ab initio model looks like. To me, it definitely is similar to the crystal structure, which comprises 80% of the structure used for cryo EM. I think especially the beta-supersandwich domain is recognizable. Please let me know what you think.
I am busy with my first ever Cryo-EM structure and unfortunately not an expert. Have you tried homogenous refinement instead of NU refine using your current particle stack and initial volume? I am just wondering if the shell you see is specific to the NU job type. Do you know where the glycans are expected?
Are you using tilt, defocus refinement, ctf refinement etc during NU refine?
I did try homogeneous refinement and the shell is there, but much less clear. But the homo ref also gives substantially worse results and terrible density maps, so it is a bit hard to determine.
Yes, we have made MS glycopeptide analysis and the position of the “density cones” makes sense.
No, I have not tried any of those settings. I will see if it makes a difference.
I think that enabling those settings may give worse results for smaller proteins so I have disabled them and only refined. But I don’t know where your shell artifact comes from. Your initial model looks good to me
Did you ever manage to refine your glycoprotein? You probably finished a long time a go but I just wanted to mention what worked for me in the end to help anyone facing a similar problem with glycans.
I first tried dilating the mask by 6 or 8 with padding of 12 during NU Refine. This seemed to work at first but then I noticed strong streaks of density near N-glycan sites. It seemed like the mask near the glycans caused overfitting there and a decrease in protein resolution. It was even worse after local refinement even when using a static mask. Dilating more (up to 20+) and way past the point of any observable glycan density, different lowpass filters, etc didn’t help. I then tried to vary all the settings in both NUR and local refine again and the *only thing that worked was to use a mask that extended just beyond the protein density (using dilation of 6 thus cutting through the N-glycans) but then padding by 20 or 30 to have a very, very soft edge. This gave me really nice refinement of the protein and good enough glycan density to allow building of the core fucosylated pentasaccharide. I’m not sure why dilating to cover the glycans caused overfitting streaks while dilating for the protein only and wide padding for the glycans worked. Maybe someone else can offer an explanation?
Thanks a lot for getting back to me! I have actually not come much further and I will definitely try your method. Just be sure, are you then just changing the “Dynamic mask near” and “Dynamic mask far” to e.g. 6 and 26, respectively? No other non-default settings?
Once I had an ab initio model that looked reasonable, I set up non-uniform refinement as below. It is dependent on the dataset though and I cannot guarantee that it’ll work for you. The padding and threshold had to be altered slightly for some of my other particle stacks (the dataset was heterogeneous) but in general low dilation and high padding worked. I also made sure that the particle stack didn’t have any duplicated particles before doing NU refine, otherwise the FSC curves didn’t drop down to zero. With local refine after this (in case you need it) I used the static mask option and created my own mask as input with similar padding as for NUR. If I used the dynamic mask option in local refine, glycan overfitting was introduced again. My alignment parameters were also set to search locally around the NUR values.
Can you please also just briefly comment on if/why the following settings helped out: “ignore tilt”, “ignore trefoil”, “ignore tetra” and “minimize over per-particle scale”
I didn’t choose to refine the higher-order aberrations as my particle is small and flexible (tried local CTF refinement before and it didn’t give good results). Tutorial: CTF Refinement - CryoSPARC Guide
In my experience, soft padding is the most important property/parameter of masks used for refinement. It is most important for local refinements, when the mask typically excludes portions of the structure and not just the solvent. When working with small masks, I’ve observed similar phenomenons as pointed out here. We’ve updated our local refinement guide page with some specific notes/suggestions on mask padding for datasets.
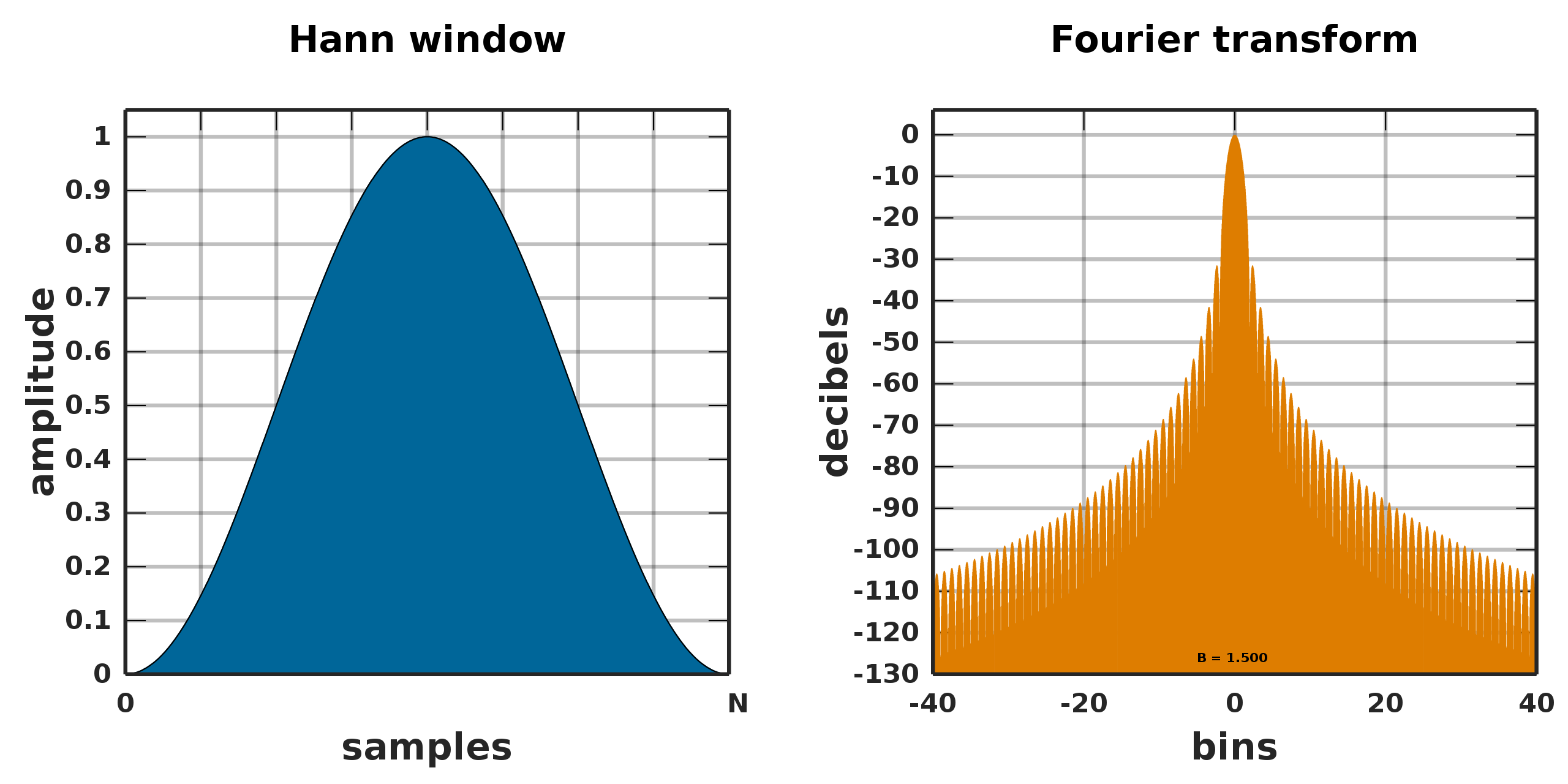
I think that in part, the underlying explanation of your observations is due to signal processing issues. If the volume is thought of as a discrete 3D signal, then the application of a mask to the volume can be thought of as windowing the signal in order to exclude regions that we are not interested (windows are applied to a signal via multiplication, just like masks). In all refinements that follow the gold-standard FSC method of regularization and resolution assessment, we must assume that the Fourier coefficients with frequency larger than the initial lowpass resolution have shared signal corrupted by independent noise. The problem with masks is that they break that last assumption – using a common mask means that the noise in both half maps (after masking) is not independent. This compromises our ability to separate signal from noise, and hence, to reduce overfitting.
Based on the convolution theorem, the severity of this violation is directly related to the Fourier-space properties of the mask. In short, the more slowly the DFT of the mask falls off over frequency, the worse the violation will be. For example, a rectangular mask (i.e. one with no soft padding, regardless of dilation) has very slow falloff in Fourier space:
The closer the mask is to a hann window (i.e. the softer the falloff in real space), the more the noise in each half-map remains independent after masking, and thus we are more able to reliably detect resolution and limit overfitting. In practice, this means that any GSFSC-based method will require trading off precision in real space (how well the masked is focused on the particular domain of interest) and precision in Fourier space (required to prevent overfitting). Heavily prioritizing real-space precision leads to overfitting and artefacts – but heavily prioritizing precision in Fourier-space means the refinement is no longer focused on a specific domain of the structure. Right now, this trade off must be considered for each refinement, but we do have a helpful rule of thumb on the local refinement job page linked above that can be used as a starting point for a good softness level.
There has been no answer on this topic for a long time, so I will try to bring up the problem of highly glycosylated proteins.
I work with rather a monomeric glycoprotein ~ 110-120 KDa, with a completely unknown PDB structure. Apart from the amino acid sequence, only the general domain structure at the sequence level is known, as to how many domains there should be. It is known that glycans constitute up to 42% of protein mass, including sialic acid, the exact locations of all glycans and their lengths are unknown.
Evidently, glycans strongly mask the protein core and so far I have not been able to visualize the secondary structures of the protein core. Depending on the 2D Classes, the Ab initio, and Refinement settings, slightly different maps are generated, including some of the settings described here.
maybe I can take a similar approach here as in this paper of Lubbe et al. (congrats - great work!) to deal with glucans.
EMBO J 2022 41(16):e110550.
doi: 10.15252/embj.2021110550
I’ve been working on it since the beginning of last summer when I got my Krios results.