It can definitely be a struggle. What non-default parameters did you use for 2D classification, ab initio and refinement? Can you share examples of 2D classes? Have you tried the new 3D classification job?
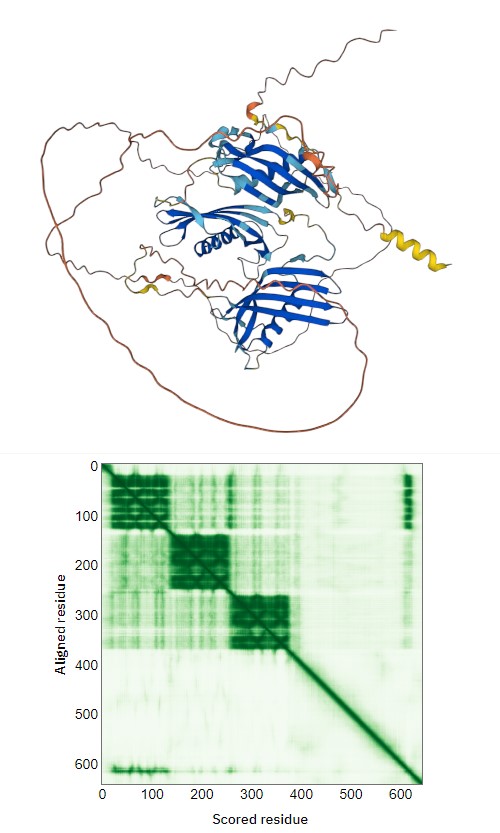
Also, can you get any leads from the Alphafold model? Does it have good confidence metrics?
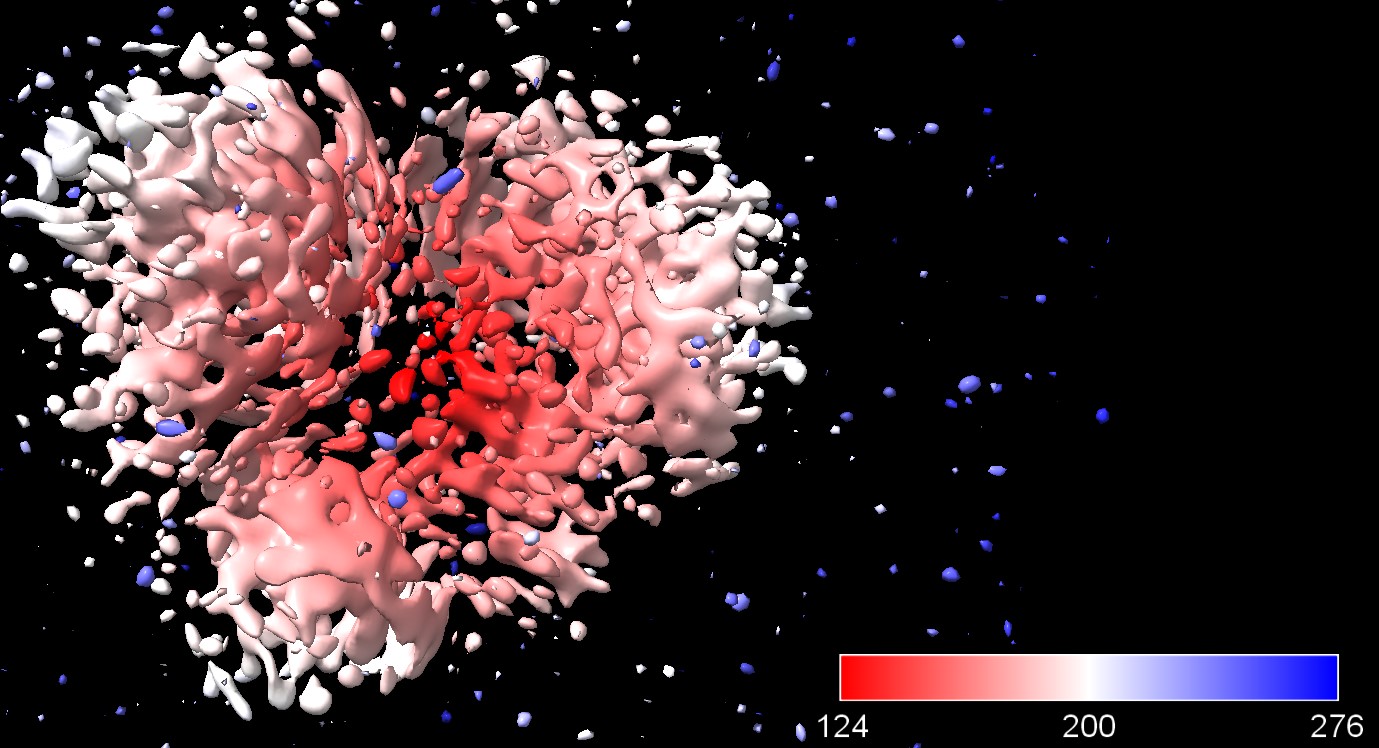
thank you for your quick response. The map images presented in the previous post (J151 & J571 were from the same particle extraction (box 256). The classification leading to the J151 (after sharpening from J149 ) was with default settings from homogenous refinement Job 149
J151 sharpen from J149: This is J149:
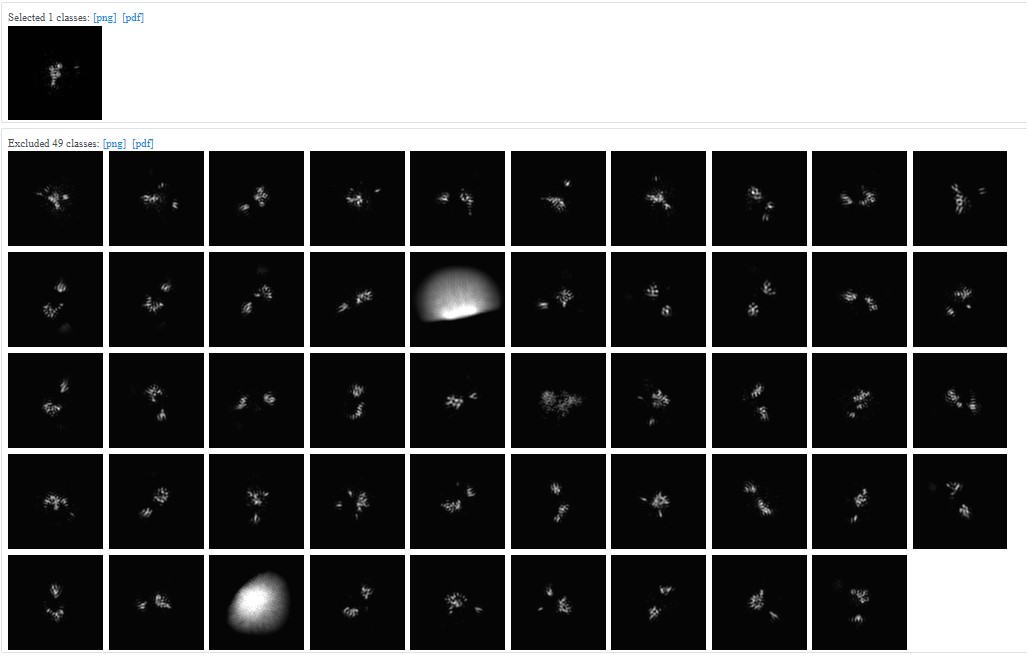
The J571 (second picture from my previous post) had force non-negative during 2D classification and only one class was chosen. Files are attached for it:
I was not doing a new 3D classification job as I remember. .
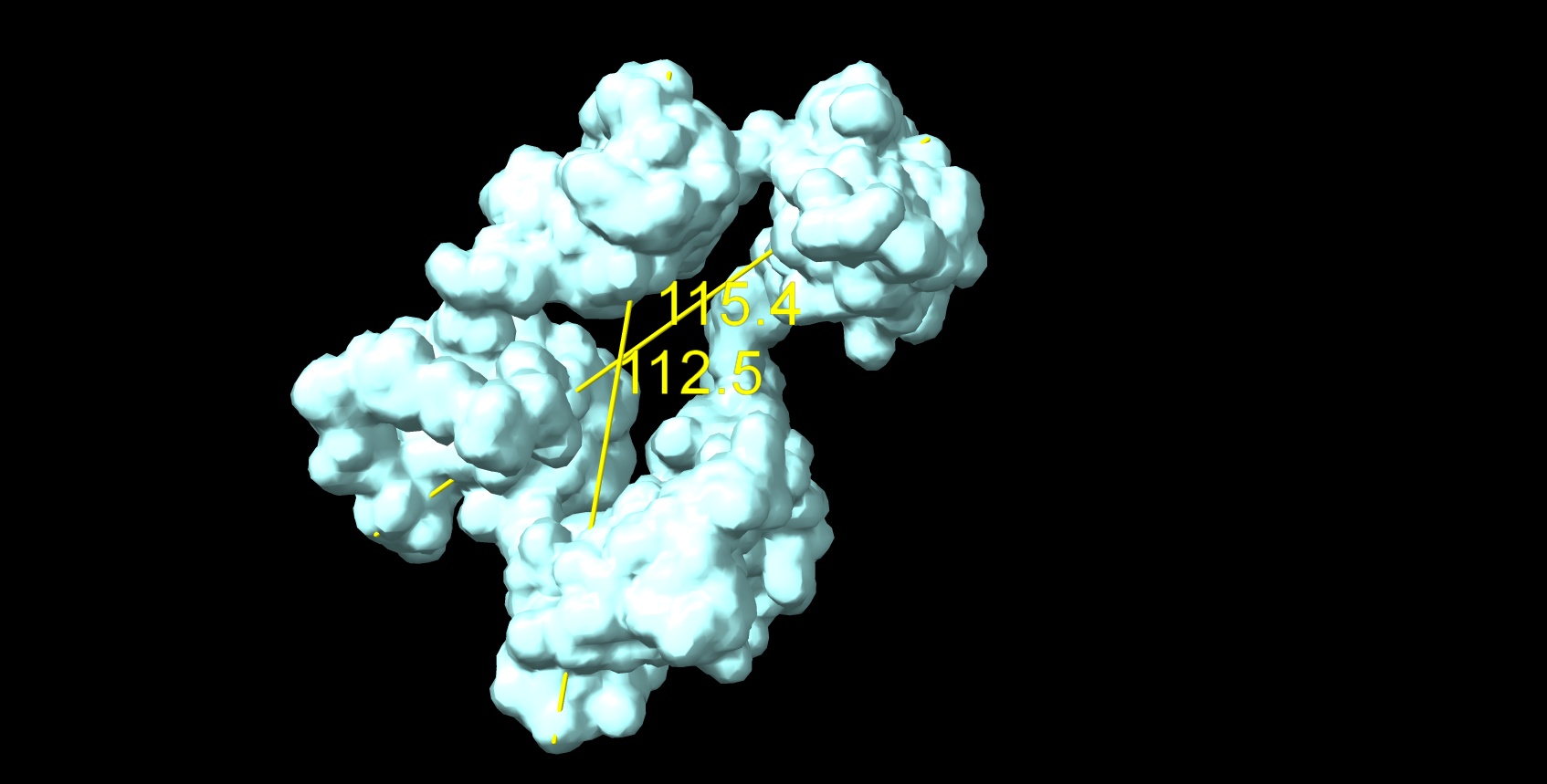
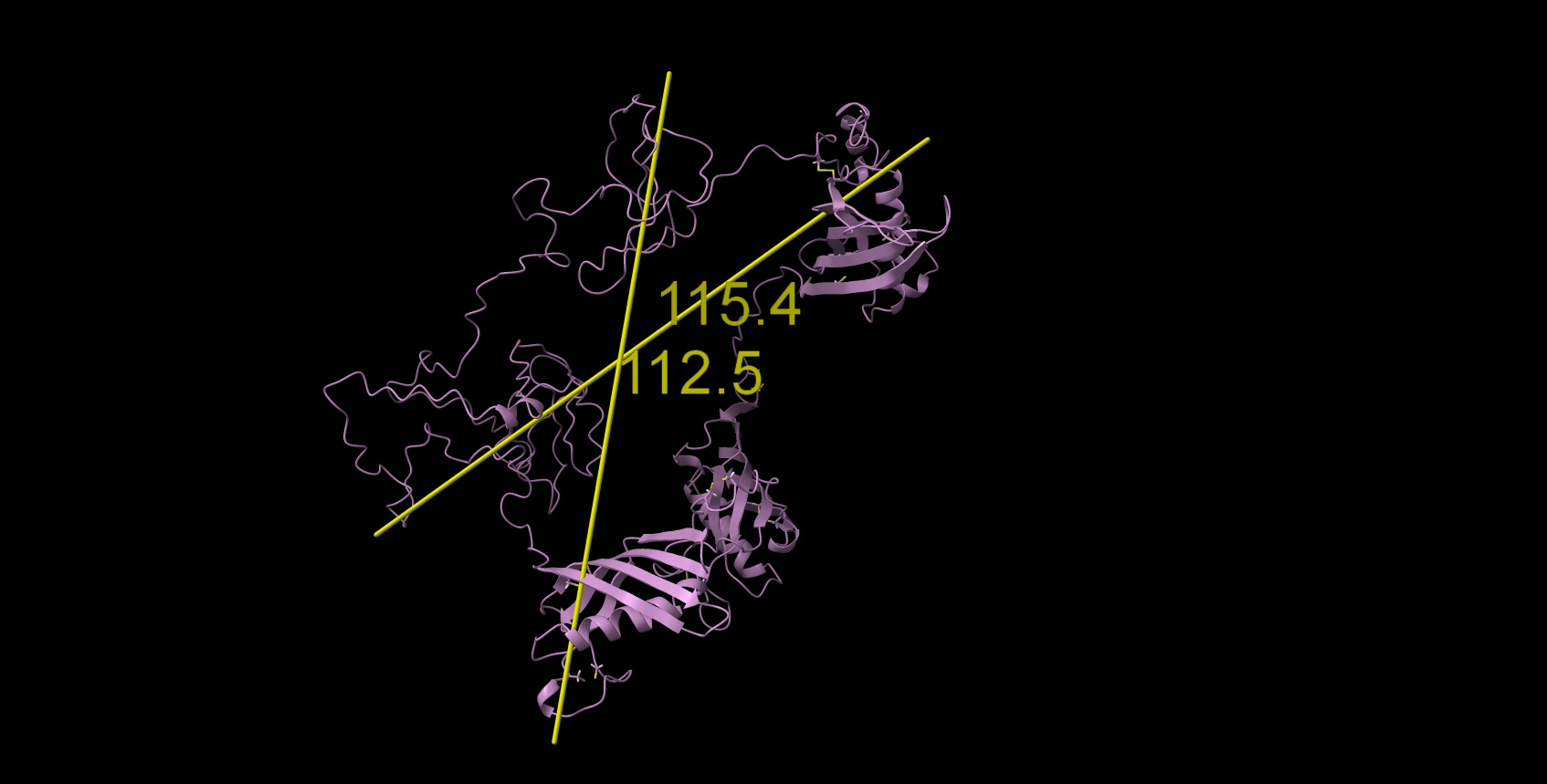
Alphafold model was able to fold only 3 first N-terminal domains with several beta-sheets and one alpha per domain (attachment, yellow N-terminal helix is redundant in native protein signal peptide), the rest is floating unfolded.
I produced some I-TASSER models of all separate domains (based on sequence fragments) and for N-terminal domains D1-D3 and they look similar to those of AlphaFold. The rest C-terminal domains are more disordered in I-TASSER, however, some have some fragments of secondary structures.
I-TASSER can’t handle all protein sequence at all to get something loong ok… Good threading templates are missing.
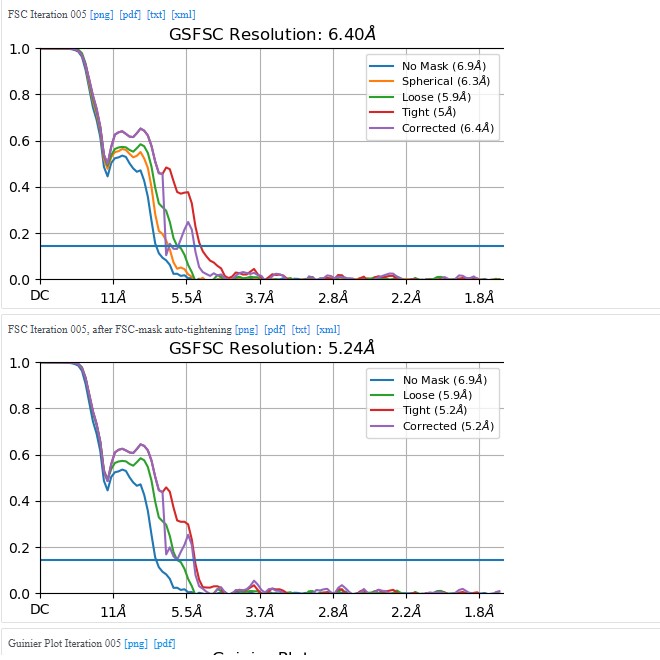
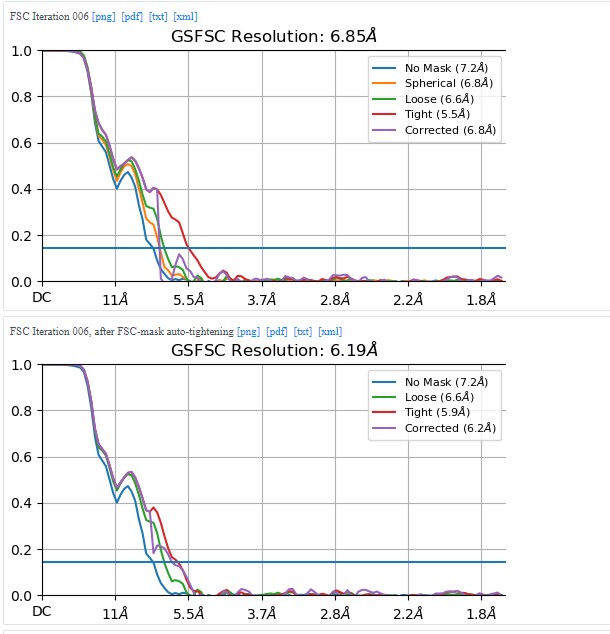
I am writing this now based on the results from cryoSPARC 3.2 saved locally. I generated a few more different maps with the setup attempts described on this topic, but as far as I can remember, none of them look any better than what is shown here.
I will try to delve into the procedure used in the publication of lizellelubbe - EMBO J 2022 41(16):e110550.and recreate it for my protein.
I’m switching slowly to the cryoSparc 4 versions, maybe there are some cool new options out there.
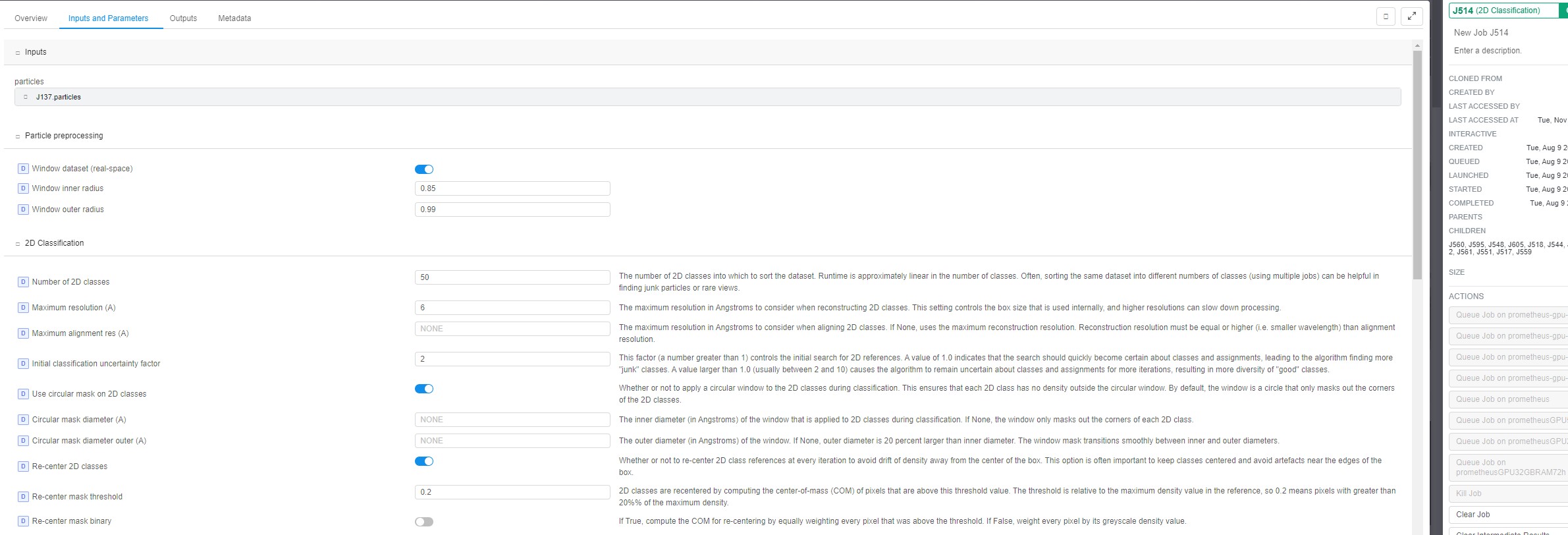
2D classification:
Keep your settings, but turn off “force max over poses/shifts” and “enforce non-negativity”. “force max over poses/shifts” off will make the job run slower, but usually yields better and more diverse classes for small proteins. Non-negativity does not seem to help you in this case, but rather creates artificial classes.
Also try using a circular mask slightly wider than your max particle diameter.
Ab initio:
Increase the maximum and initial resolution to something like 6/15 Å. This will make the job take significantly longer to finish, but is sometimes necessary to yield reasonable ab initio structures of small proteins without larger distinguishable features.
Also increase the initial and final minibatch size to 300/1000. I’ve sometimes increased to as much as 1000/5000, but then the job takes forever.
Refinement:
Decrease the “initial lowpass filtering” to something like 12 Å to “keep” more features from the ab initio structure.
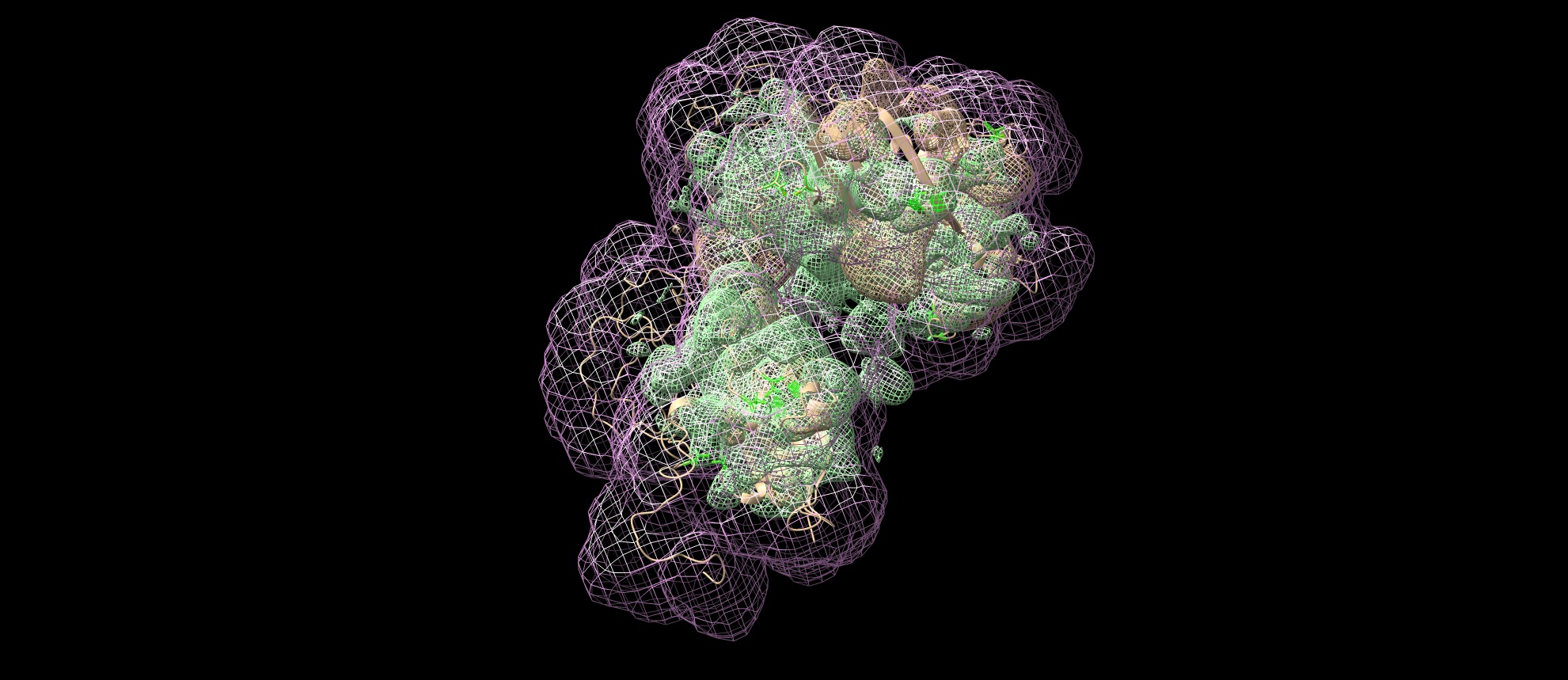
Here is the Chimera-generated map (min max level) of my protein prediction model, partially based on AlphaFold (3 good domains) and iTasser (the remaining domains which are predicted to have no secondary structures and are the most highly glycosylated - the most known sites described in UniProt. The entire structure should form a beaded necklace because there is a disulfide bridge between domains 1 and 6. I produce it by joining models from the domains in Chimera and optimized these interdomain connections in Foldit Standalone. The Ramachandran chart looks good.
The maps were fitted by shape because the secondary structures are still not visible even for those domains well folded in beta sheets and 1 alpha helix, so there is not much to match exactly. I think I need to filter the particles better, but before that, I can do motion correction MotionCor2 and all over again picking several times to filter the particles. The current results are from patch motion.