Hi CryoSPARC folks, I’m having trouble with getting good non-uniform refinement results. The HR-HAIR (i.e. Ab Initio) method proposed by @olibclarke gets me to a decent starting point, but when I switch to non-uniform refinement, things start falling apart. I’ll elaborate on the current workflow. Thank you all for any wisdom/advice!
Using Relion, I was able to pick a decent set of particles that appears to have my protein complex of interest. There are 3 components: a dimer binds a monomer. You can see 3 clear lobes in the Relion 3D classification.
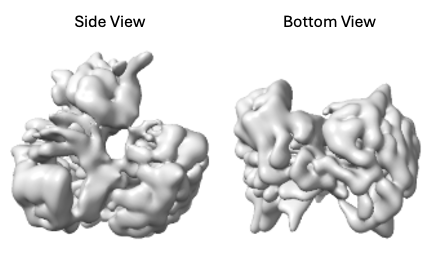
Then, I performed 3D auto-refine in Relion, which gets me around ~6 Å GFSC gold-standard resolution with no obvious severe preferential orientation issues.
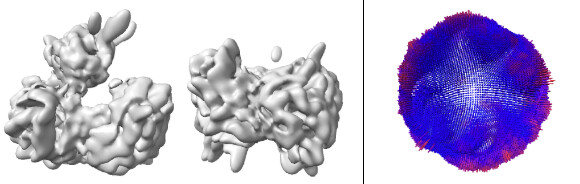
After 3D auto-refine, the CryoSPARC work begins. I extracted these particles from Relion (3x particle diameter box size) and imported them into CryoSPARC. Here are the steps taken so far in CryoSPARC:
-
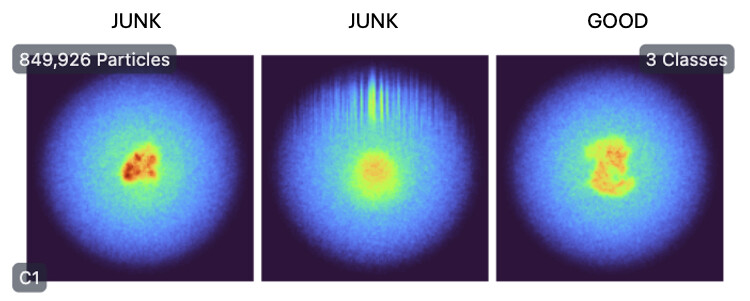
HR-HAIR run with 3 classes. Unfortunately, the job did not fully finish, due to issues detailed HERE (would appreciate any help for this issue too). The job was able to pull out a decent amount of junk though, which is promising.
-
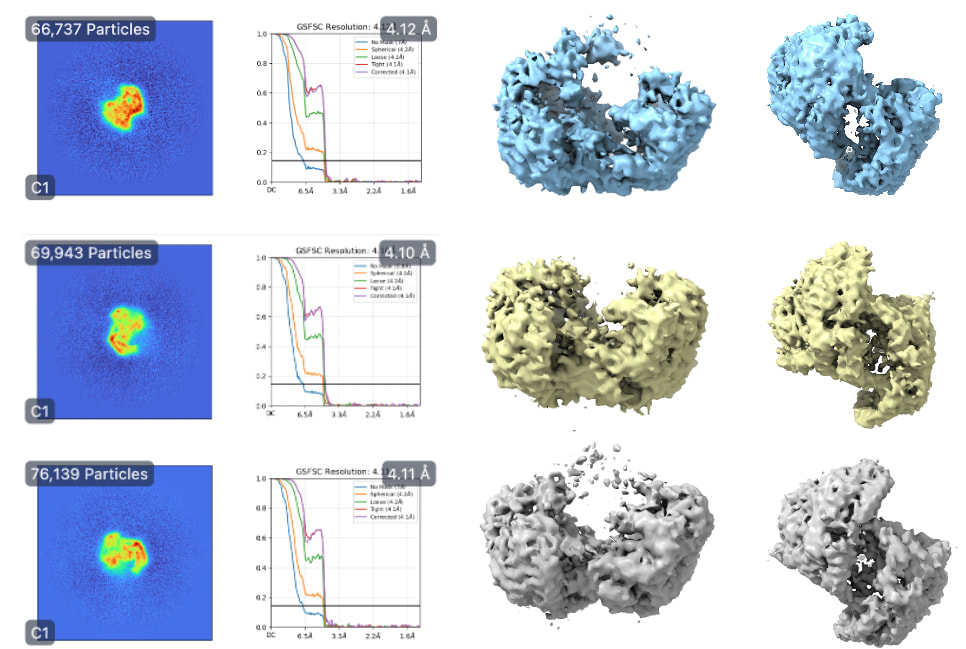
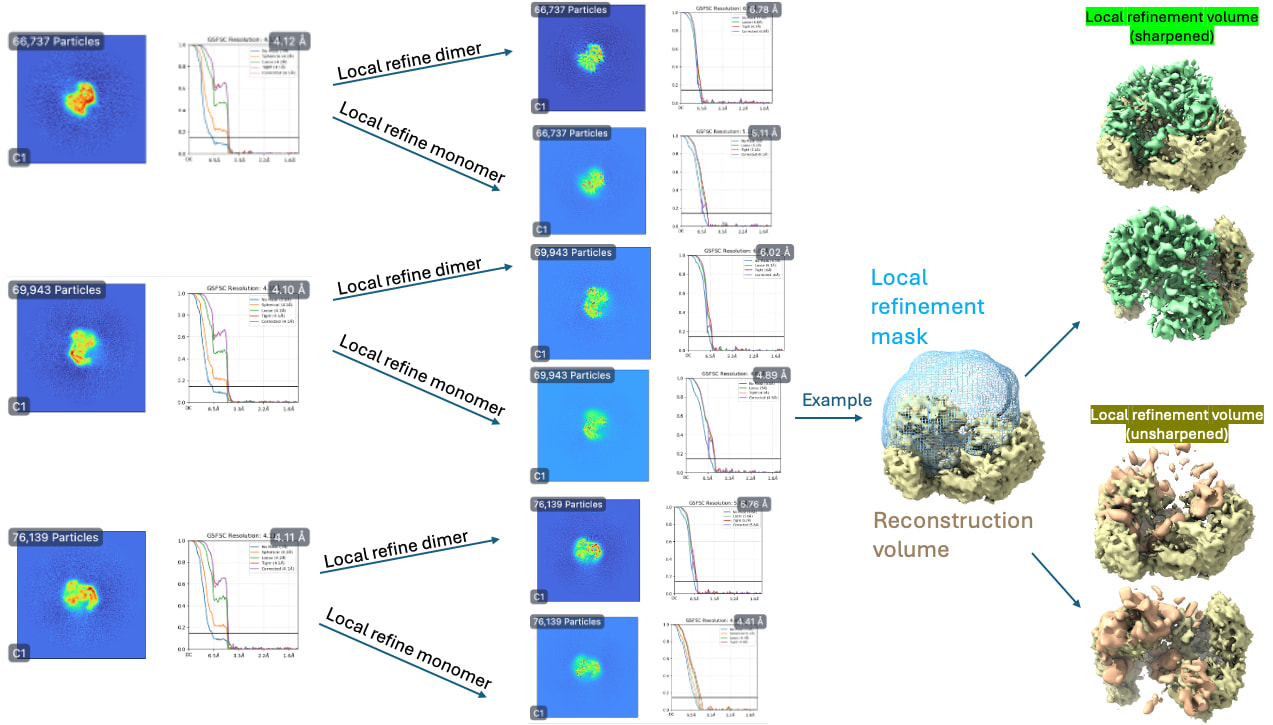
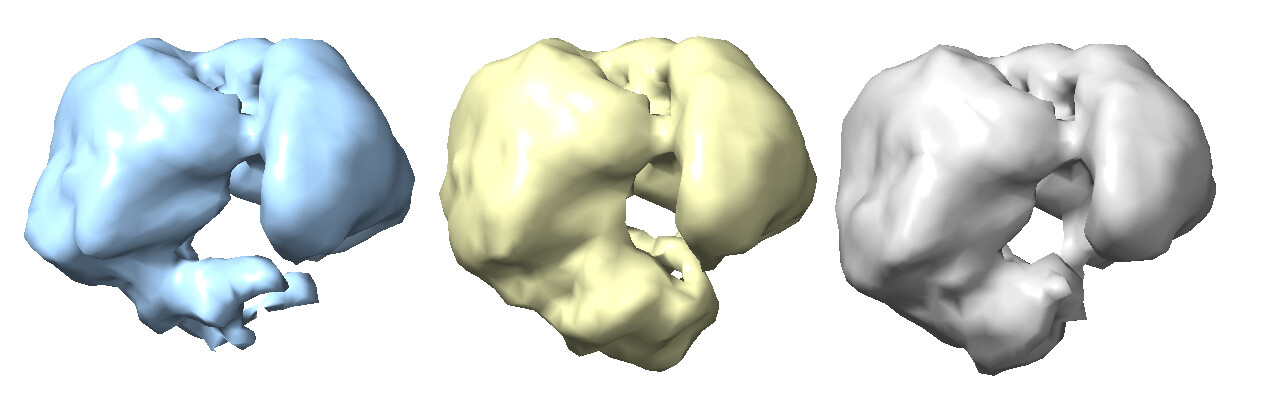
Subsequently, homogenous refinement and non-uniform refinement jobs on Class 2 volume and particles (the “GOOD” class). This is where things seem to be off. The reported GFSC resolutions don’t match to the features I should see. The density also seems very “blurry.”
-
I figured the refinement jobs might not have worked well because there was too much junk still. So, I took those particles into heterogenous refinement to pull out more junk.
-
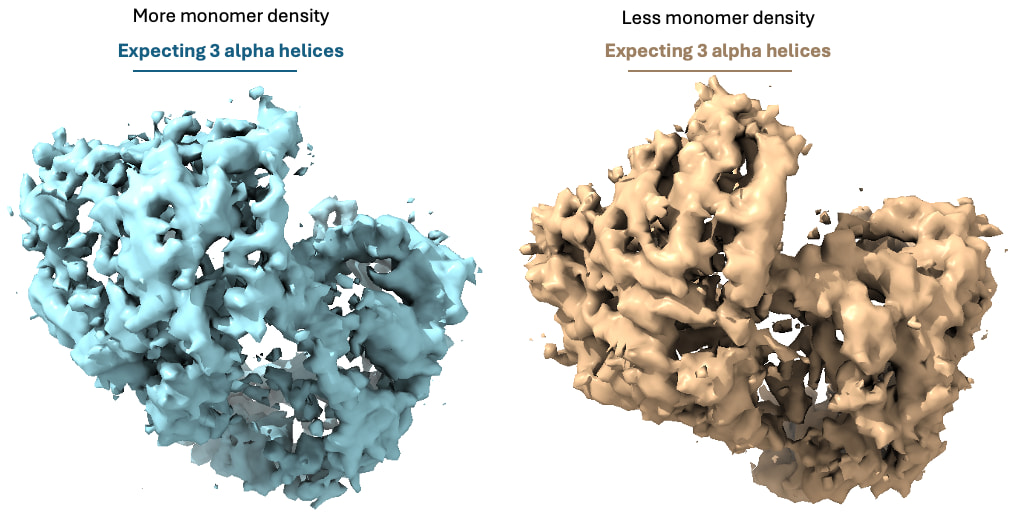
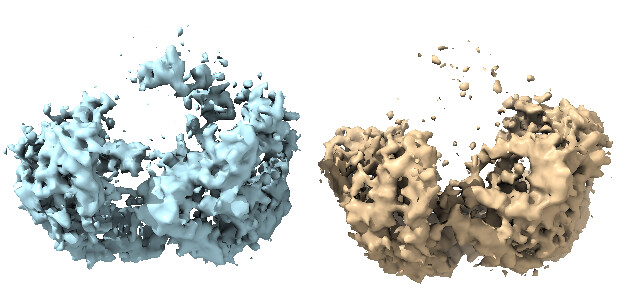
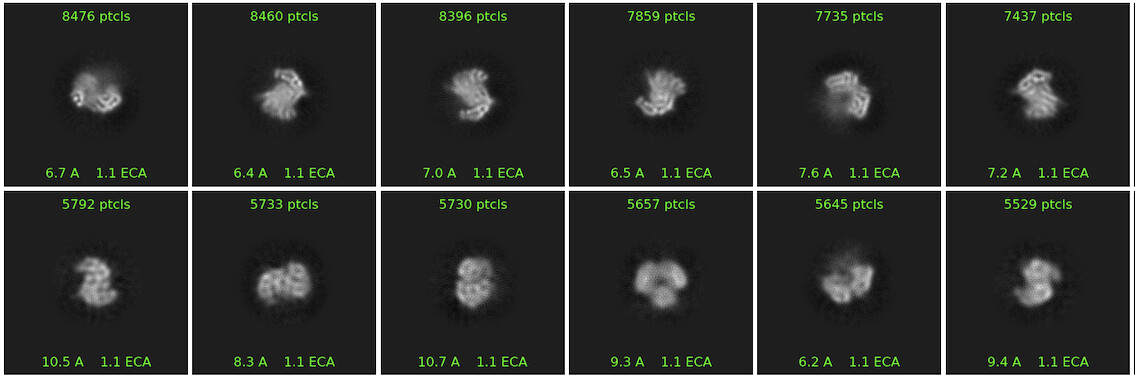
Then, I went back to HR-HAIR with ~213k “GOOD” particles, again with issues running the full job, but I am starting to see features of the already published dimer (without monomer bound). This was really exciting because I’m seeing alpha helices exactly where I expect them. Something interesting was that I noticed that, in later iterations of HR-HAIR, the monomer “disappears,” but the expected features become more prominent. I am thinking that HR-HAIR does a good job at classifying the “rigid” part of the complex, but the monomer might be more flexible/dissociate, leading to weaker density. I also observed this in another diagnostic 2D classification run.
-
However, this is where things start to get weird again… With this theoretically better set of particles, I took the best volume of HR-HAIR and ran non-uniform refinement. I am doing this because I’d like to eventually go to local refinement, where I can hopefully try to pull out the monomer with weaker density. Unfortunately, I am observing strange non-uniform refinement behavior like in Step #2: blobby density that does not correlate to the reported GFSC resolution.
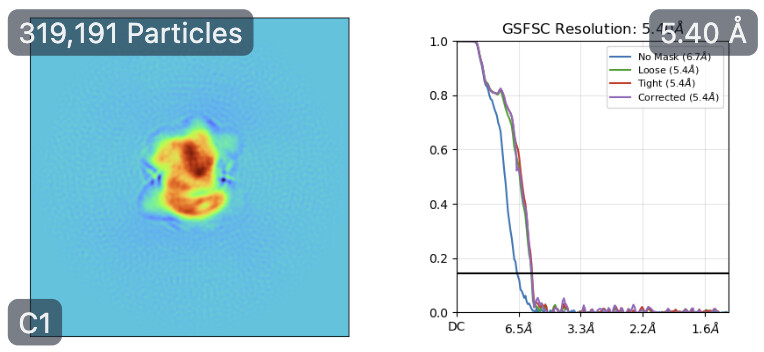

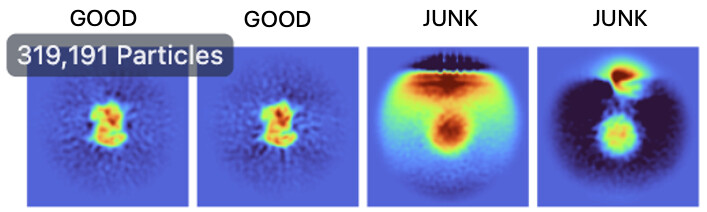
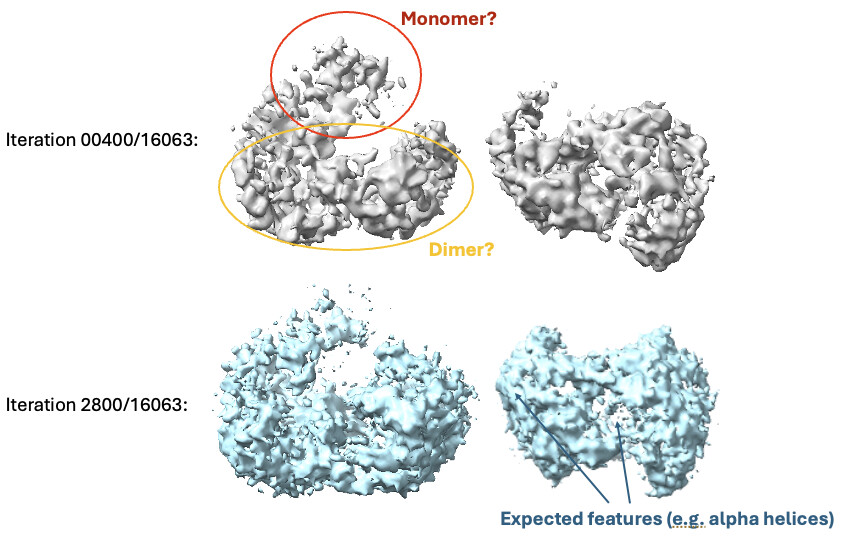

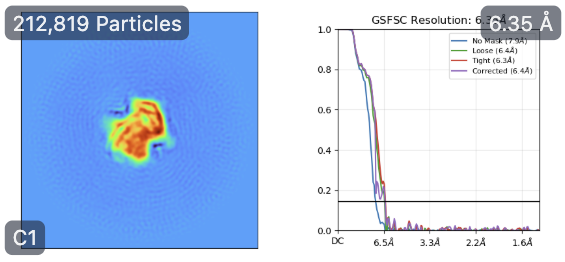

Any thoughts on what’s going wrong here once I take things into refinement jobs? HR-HAIR is clearly getting me somewhere, and it would be great if I can really start locally refining that monomer density. My hunch is that the monomer is bound/unbound and might have a lot of flexibility, which could throw these algorithms off. Does anyone have a suggested workflow that can help me push this dataset further? It might be impossible to get a low-resolution map with both components, but maybe I can get each individual component (dimer and monomer) locally refined well enough that I can create a composite map to make an atomic model.
Thank you all for your advice.