hi,
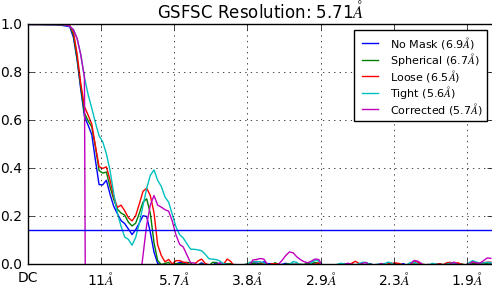
when I finished the homogeneous refinement I got the GSFSC like this, can somebody help me what wrong with it and what I should do next? if I do heterogeneous refinement with this output map, the resolution down to 8A, I do’t know what’s wrong.
Do you see any features in the map that look like helices (assuming your protein does have them)? Also, what would happen if you use the particles from each of your hetero refine job and do homogeneous refinement again?
One other thing you might consider trying is to play with particle extraction pixel sizes. Changing the box sized up and down may allow the program to find the protein features easier. To re-extract particles with different size, you only need to drag the good particle collection from your refinement job output and the micrographs from the ctf job into an extraction job - no need to go back to the original picking.
This sort of FSC is due to masking artefacts caused by the mask generated by the FSC calculation algorithm. You would get the same artefact in RELION if you provided a mask that was too tight. RELION provides a warning message when you get that, however cryosparc does not.
A solution can be to provide a mask manually or to drop the threshold for the dynamic masking. I have gone as low as 0.05. A good idea would be to look at your map.mrc file as well as the mask files (both mask_refine and mask_fsc) and see if the masks are cutting your protein density.
I believe my problem is disordered protein density at the surface of my molecule. Could this be an issue for you also?
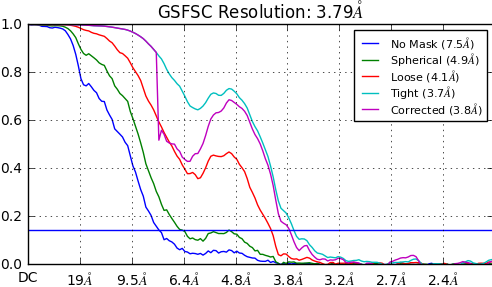
Followed by this topic, I’m curious about the tolerance of the dip from the FSC corrected curve. sometimes we see the corrected curve even dip under zero then we know we need to do more, but sometimes we see only a small head down at high resolution or deeper like this attached file.
So is there a good rule of thumb like to which extent can we publish it ?
Is this a membrane protein? Membrane proteins commonly have a dip in that region, due to some influence of the micelle. For a very high resolution membrane protein which is cleanly classified, the dip will get smaller, but usually doesn’t go away entirely.
As mentioned previously, too tight masks can also cause these dips - I would be especially careful about that since you see such a big difference between the spherical and loose mask.
If you have a very sharp dip at higher resolutions than what you see here, like around 2 - 4, than defocus refinement may help.
Thanks! Very informative. You’re right, It is a membrane protein, we know it’s common for membrane protein to have this dip. Seems in my case it mainly affect by the mask provided. Could you explain more about why the big difference between spherical and loose mask rise more concern? I would simply think this difference is caused by the way the mask was generated. What else could cause this difference? Thanks!
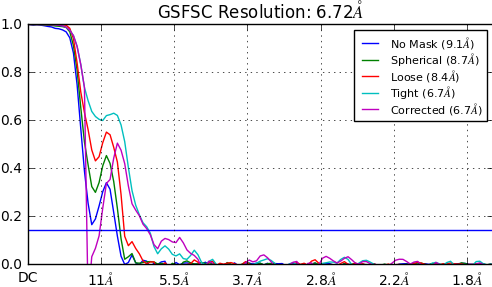
I am experiencing the same problem, but the protein I am working with is not a membrane protein. However, it is fairly small (100 kDa), monomeric without symmetry and carries four N-glycans. Do you know if flexible glycans might also cause this kind of behaviour?
There’s probably a lot of stuff outside of the mask, which is making the corrected curve drop so sharply. Try doing a regular refinement with the mask resolution set to like 1A so there is no mask. It may help figure out how to direct further refinement. Also try the non-uniform refinement.
I got a GSFSC curve like this. This is a membrane protein. Your idea must be right. It because the influence of the micelle. Could you give more information about this influence? How does the micelle cause a dip? Thanks a lot!!!