I am fairly new at cryoEM and have been using cryosparc for my reconstructions on a dataset. I was hoping for some quick comments/advice/recommendations from experienced users of cryosparc/cryoEM experts.
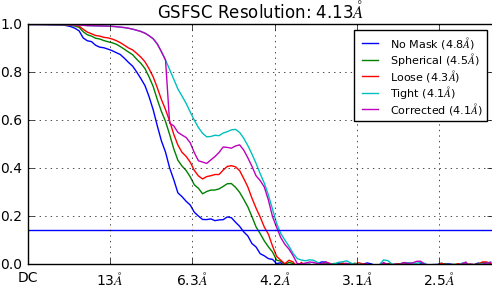
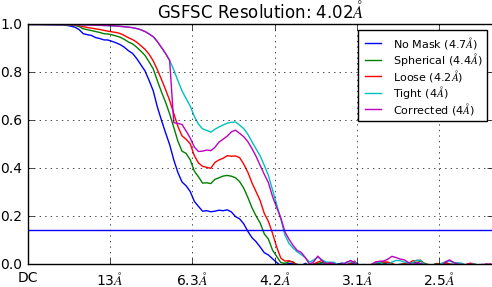
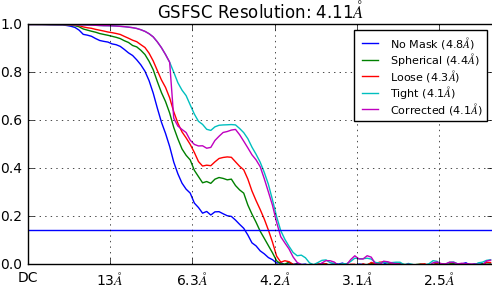
For 3 different subsets of articles from the same full dataset, I get reconstructions between 4.02-4.13A. The maps overall seem to be comparable - some parts of the protein are clearly at high resolutions with clear side chain densities but some parts are at far low resolution (5.5-7A range). I get the following FSC curves for the 3 different maps (see below). The tight and corrected curves seem to converge in the high resolution range but seem to deviate in the low resolution range. My questions are:
(1) Do you think my reconstructions would be trustworthy given these FSC curves?
(2) Would I be correct in concluding that given these FSC curves, the features in the 5.5-7A are not trustworthy? (these low resolution features correspond to important domains of my protein - I am wondering whether it would be technically correct to dock homology models of the domains into the densities to identify how these domains are organized in the full protein. I understand that trying to build in models into these low density features would be risky/inaccurate but with respect to domain organization would it be okay to use these low resolution features)
(3) Given these FSC curves, would you consider the maps to be at publication level? (That would probably be hard to answer I know. But I am concerned whether there is something glaringly incorrect in my FSC curves which would clearly point towards something wrong. My maps seem to be fairly reasonable as far as I can tell.)
Any tips on what tests I could do or what I should look out for in maps to be sure that “everything is ok”.
Maybe someone more expert than I will chime in, but here’s my non-expert reply.
The dip in the corrected FSC is normal. It’s where phase randomization begins in the calculation of the corrected FSC. You will always see this.
In my experience, if the dip is very large, it can indicate potential problems, such as duplicate particles, or masking problems. I’ve encountered both problems, and when I corrected them the dip was smaller that what you’re showing, although what you show is not that bad and probably usable.
Some suggestions:
Note the B-factor from the Guinier plot of your refinement, and use the refinement volume and mask output as the input for a Local Resolution job. Then use the output volume of the Local Resolution Job as input for a Local Filtering job, and enter the noted B-factor for sharpening (as a negative number). This will improve your map. You can also download the LocRes volume from the Local Resolution job, and the output of the Local Filtering job and display it within Chimera using Volume->Surface Color to color the filtered map according to the local resolution. That, plus your eye will tell you what you can confidently model and what you can’t.
Run a Heterogeneous refinement to look for different populations. How much you can get out of this will depend on how many particles you have to work with (and how variable your protein is).
Run a 3D variability job to look for domain movement. If you identify major movement from this or the hetero-refinenent, you can run a Local Refinement to improve the resolution in the area of interest.
Vow! Thank you @rj.edwards Your suggestion #1 was wonderful - it makes a dramatic improvement to my map quality.
I have tried heterogenous refinement into ~3 classes and the 3 classes separately refine to a resolution lower than the original map. The maps however do not appear to be much different from one another as far as I can tell (159K particles give me a 4.02-4.14A map and the sub-classes, ~40-60K give me 4.4-4.6A maps)
I will try 3D variability!
Thanks a lot for your recommendations! (Always more things to try :))