Hi  ,
,
I’m facing more or less the same problem. I’ve collected a dataset of a small and elongated protein complex (~75 kDa) with good contrast too. We align the 2D but we have issues with the 3D.
I attach a pair of micrograps for reference:

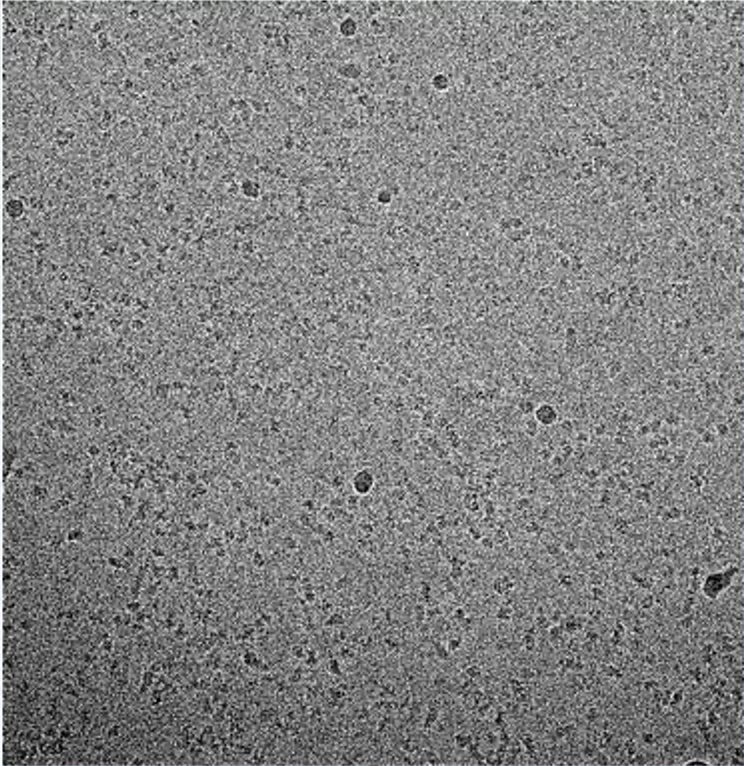
They were collected on a Talos Arctica with a K2 at 36 K magnification (1.2 A practical, 1.13 A corrected). One shot per hole on a 1.2/1.3 Au grids. Defocus was settled between 0.5 and 1.7 um. The dose is 50 over 65 frames (0.77 e/A/frame). I curate the micrograph to select the ones with ctf fits better than 5 A and I extracted around 2 million particles.
I use previous data to pick particles and I extracted them with a 320 box size.
I run a 2D classification and I get good 2D classes (image after several 2D classification to clean the dataset):
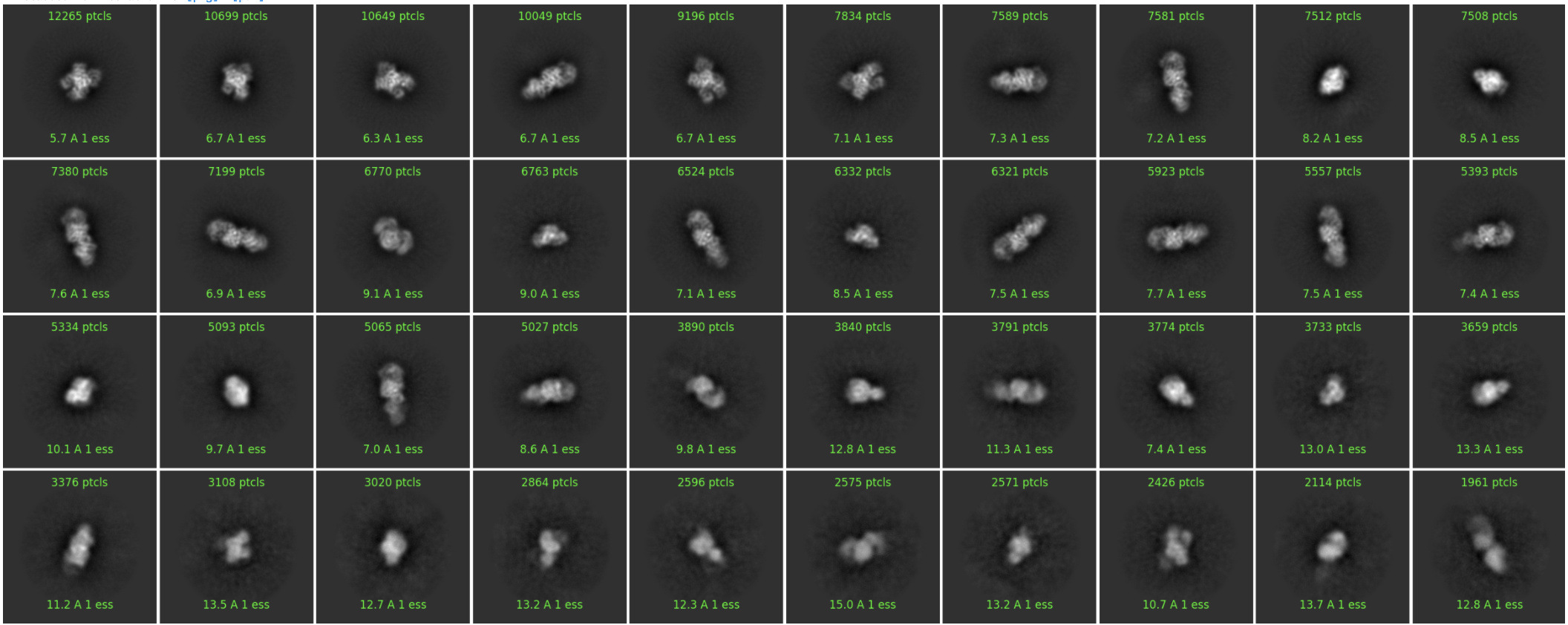
*Even though, I was trying to use the parameters described here to maybe improve my 2D classes (I’ll explain that after).
But then, when I move to the Ab-initio and try the 3D reconstruction (1, 2 and 3 classes) I don’t get good models.
1class
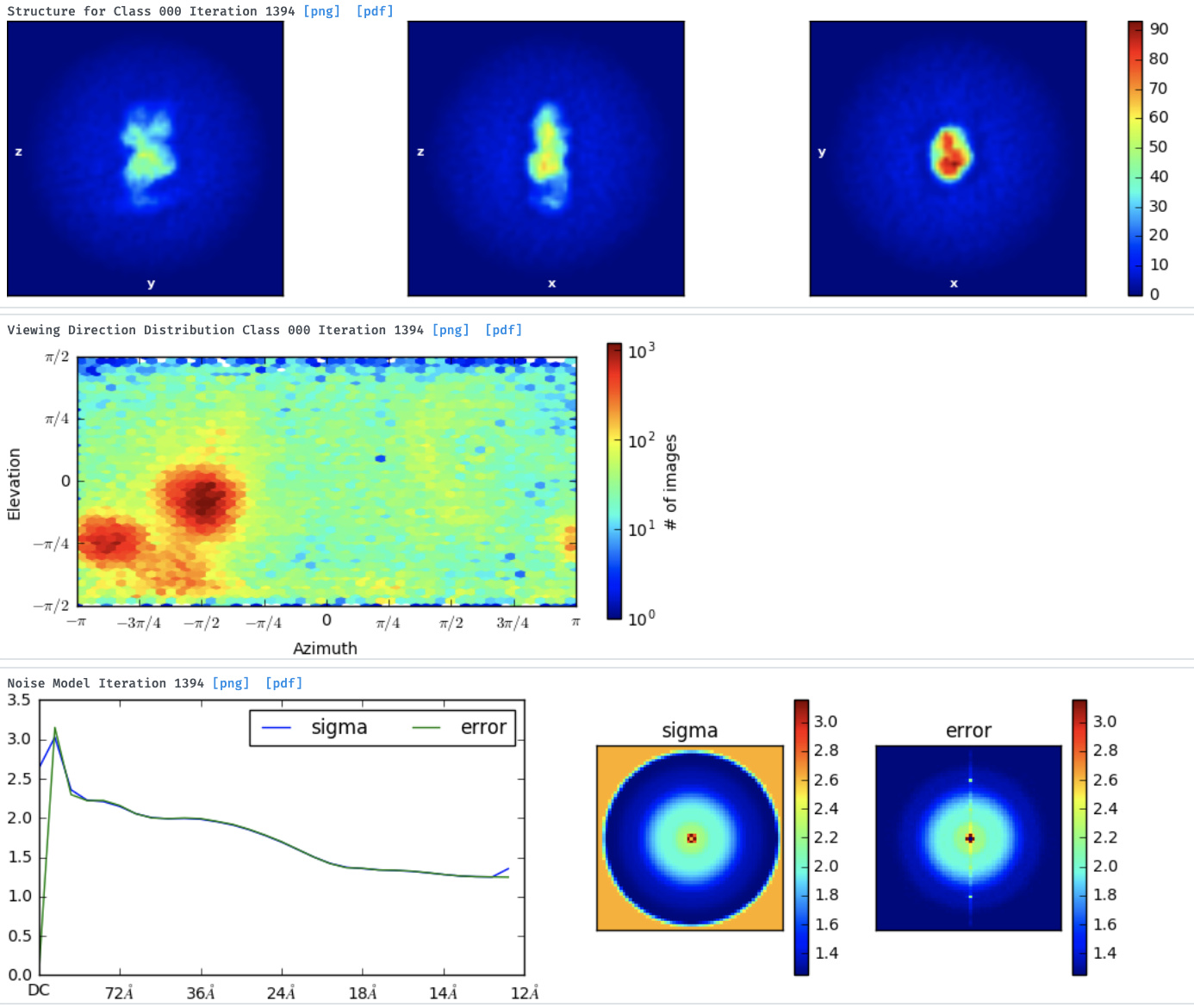
Either with the NU-ref.
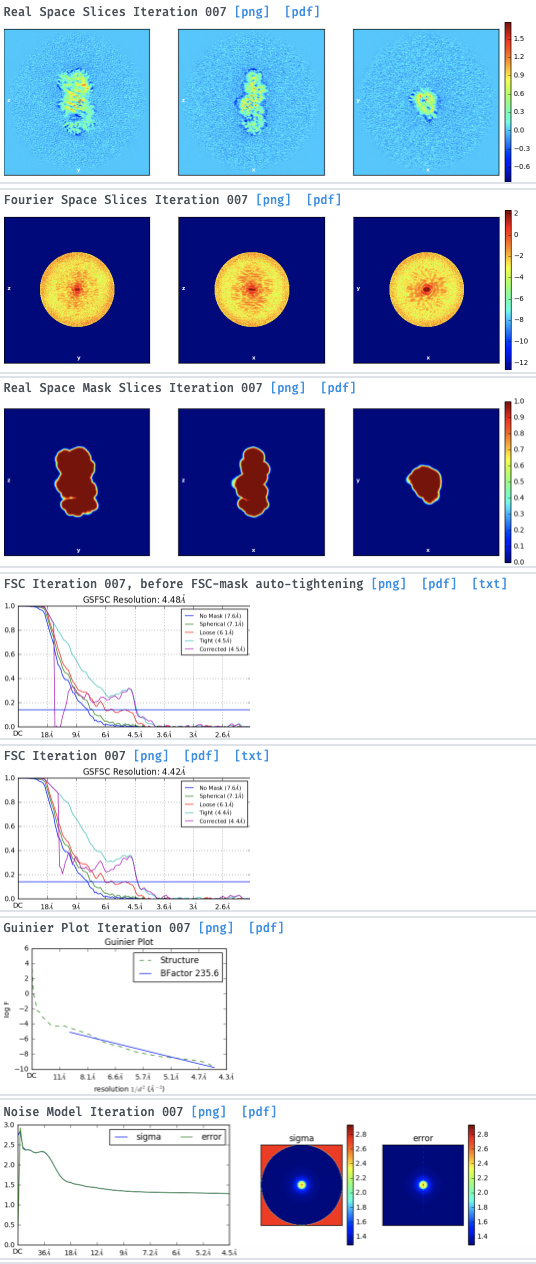
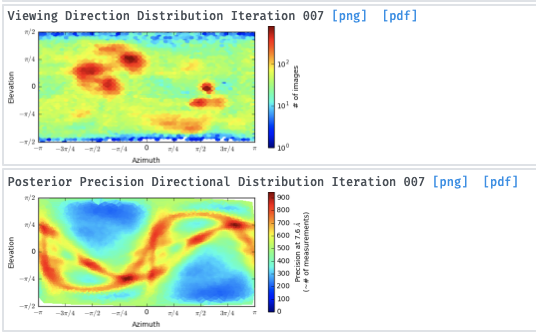
Even if the particles are small I think the 2D classes are kind of promising and I think it could be possible to improve the 3D model to get a good one. So, I tried the advises that are proposed here but for both 2D and 3D, i get the same error:
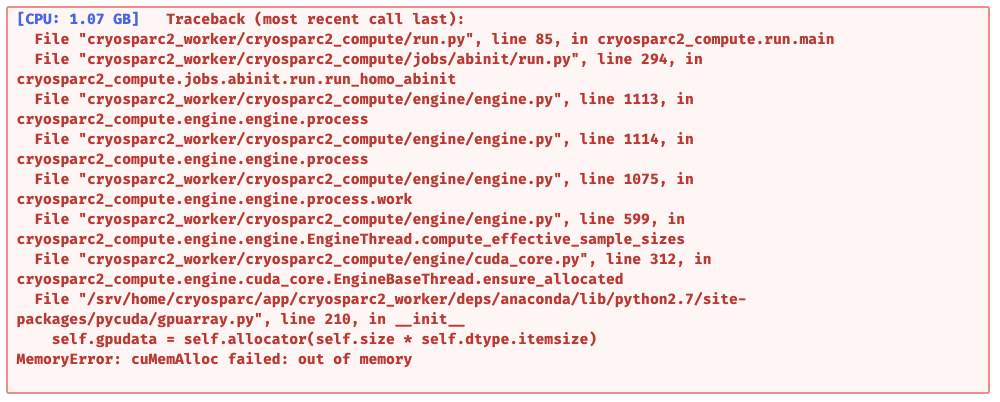
I understand it’s a lack of GPU problem, so maybe my job settings are too exigent or they make non-sense at some point, or maybe both.
My 2D parameters are:
- Number of 2D classes: 80
- Max resolution: 3
- Initial classification uncertainty factor: 4
- Re-center mask threshold: 0.1
- Re-center mask binary: ON
- Force Max overr poses/shifts: OFF
- Number of online-EM interations: 40
- Batchsize per class: 300
- Number of iteration to anneal sigma: 25
- Cache particle images on SSD: OFF
And using my previous 2D classes I was trying to run an Ab-initio job with the following parameters:
- Maximum resolution (Angstroms): 9 (before I tried 7)
- Initial resolution (Angstroms): 12 (before I tried 9)
- Initial minibatch size: 400
- Final minibatch size: 1200
- Cache particle images on SSD: OFF
- (For 1, 2 and 3 classes).
If someone have any suggestions or ideas I’d be really happy to try them .
Lu.