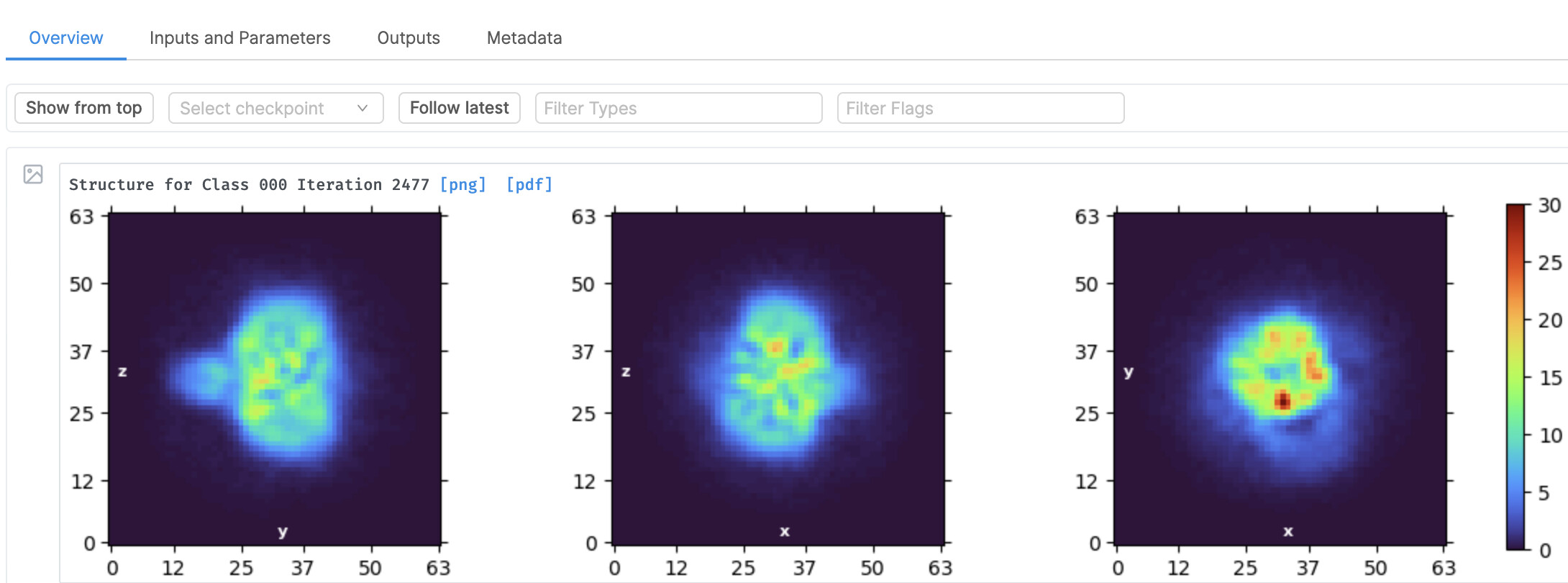
The initial model (ab-initio) protocols create quite a nice initial 3D map, but any further 3D refinements(homogeneous//non-uniform…) make the 3D map very bad.
Does anyone have the same expertise, and does anyone know the possible solution?
If this is a small protein, masking in non-uniform or homogeneous refinement may be causing issues. Try using non-uniform refinement, and switching masking off (set dynamic mask threshold to -10, or set dynamic masking resolution to e.g. 1Å)
Try local refinement starting from the ab initio map and particle set. If refinement is not converging, sometimes this helps.
how many particles? also try: 1) redo ab initio with multiple models, hopefully you get the same result but simultaneously also get rid of particles that do not fit the model (in the other classes). 2) perform classification in 3D with heterogeneous classification for the same effect.
We also tried to bin the particles and run them in the general routine, but that did not help.
Also, relion produces similar results (with a better b-factor).
redo ab initio with multiple models, hopefully you get the same result but simultaneously also get rid of particles that do not fit the model (in the other classes). yes we tried that - and the results look good for ab-initio but terrible for any 3D refinement
perform classification in 3D with heterogeneous classification for the same effect. Yes, we also tried that - it works (splitting works nicely) but again, just till 3D refinement.
Are you sure the ab initio is a correct solution? To me it looks a little like one of the failure modes we often see, described here: "side view prior" for ab initio?
It doesn’t seem to have many high resolution features - can you clearly see secondary structure in the ab initio, and what settings are you using for ab initio? Are clear secondary structure features present in the 2D classes?
Good question – from data that I have – perhaps, yes. But after you mentioned that – I am intensively checking and remastering it. Thank you for your remarks.
Did you find a solution at the end? As I understood, it happened due to limited views. Did the re-picking step help?
It doesn’t seem to have many high resolution features - can you clearly see secondary structure in the ab initio, and what settings are you using for ab initio?
Well, not really – I see a bit in 2D classes. The setting is the default ones + 0.01 for the Fourier redius step (I found it worked better for me)
Are clear secondary structure features present in the 2D classes? These are the ones.
In some cases repicking helps, in other cases altering parameters for ab initio (resolution range in particular). Sometimes if doing a multi class ab initio, some classes will fail like this, where others give the correct solution.
These look decent. Looking at these, I would guess your protein has Cn symmetry (probably C2 or C4) - it may be worth experimenting with enforcing symmetry during ab initio and refinement.
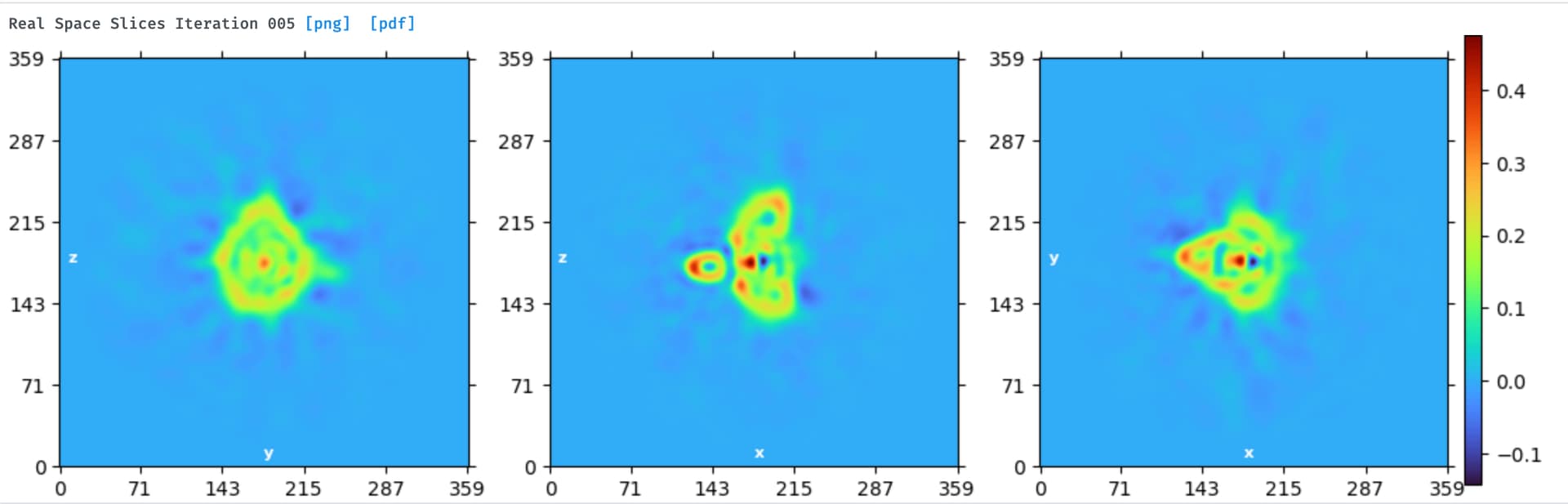
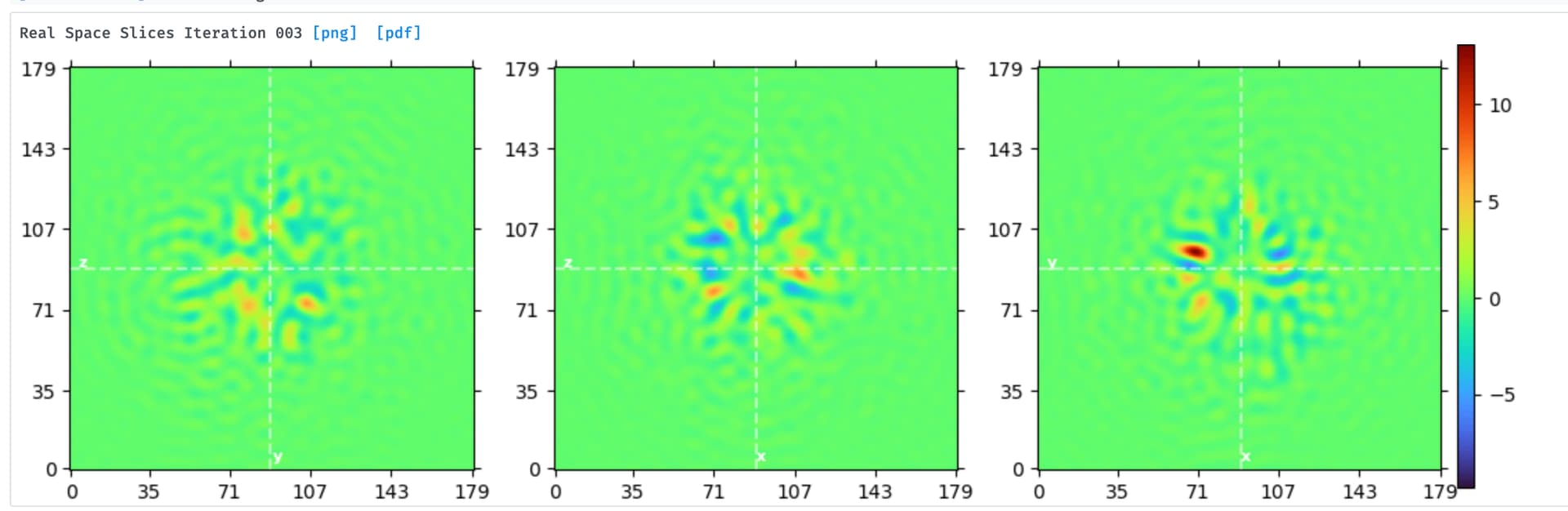
Hi Dmitry, I met same question seems like what you refered:
good ab-initio
one good class in hetero-refinement
and bad NU-refinement
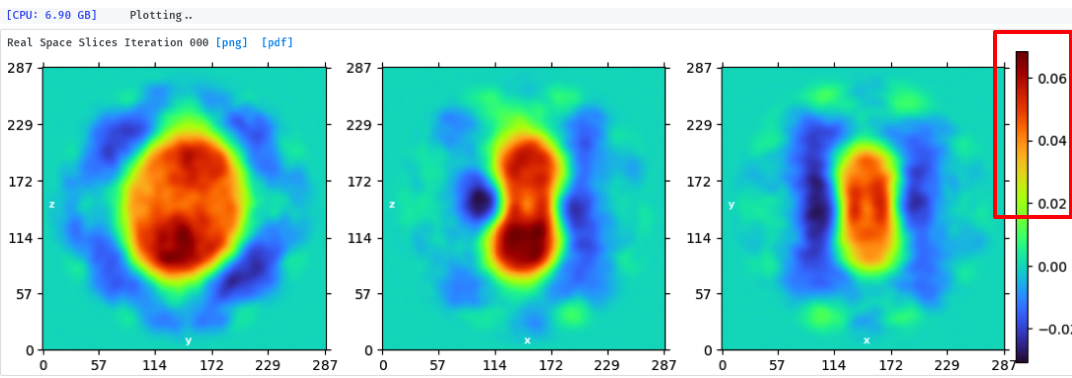
And @olibclarke 's answer about mask gave me a hint.
I checked the intermediate results of first 3 rounds of my bad NU-refinement and found that the density of region of interest is just over 0.05 (base on the distribution bar on the right), which is smaller than the default "Dynamic mask threshold“ (0.2). So I changed that initial value of mask generation to 0.05 and the result seems good now.