Hi,
I have a consensus refinement of a glycoprotein to 4.34A global resolution and used it as input for 3DVA with a lowpass filter of 8A and 3 modes.
The motions I observed are large glycan motions, flexing at a hinge point between two subdomains, and breathing within each subdomain. I find this very interesting and would really like to observe it by reconstructing the individual states using NU-refine. Since it is continuous, 3DVA clustering doesn’t seem to be an option for creating particle subsets for downstream refinement. I have seen this post ( Continuous vs Discrete motion in 3DVA - 3D Variability Analysis - cryoSPARC Discuss) but am still not clear on how I can further analyze the motions I observe. Would local refinement with a mask around each subdomain be a good idea? Or could ab initio and hetero refine be used to separate these differences?
Due to the small size (140kDa) and high flexibility, I am not sure if local refinement will be possible after particle subtraction. I would really appreciate any advice!
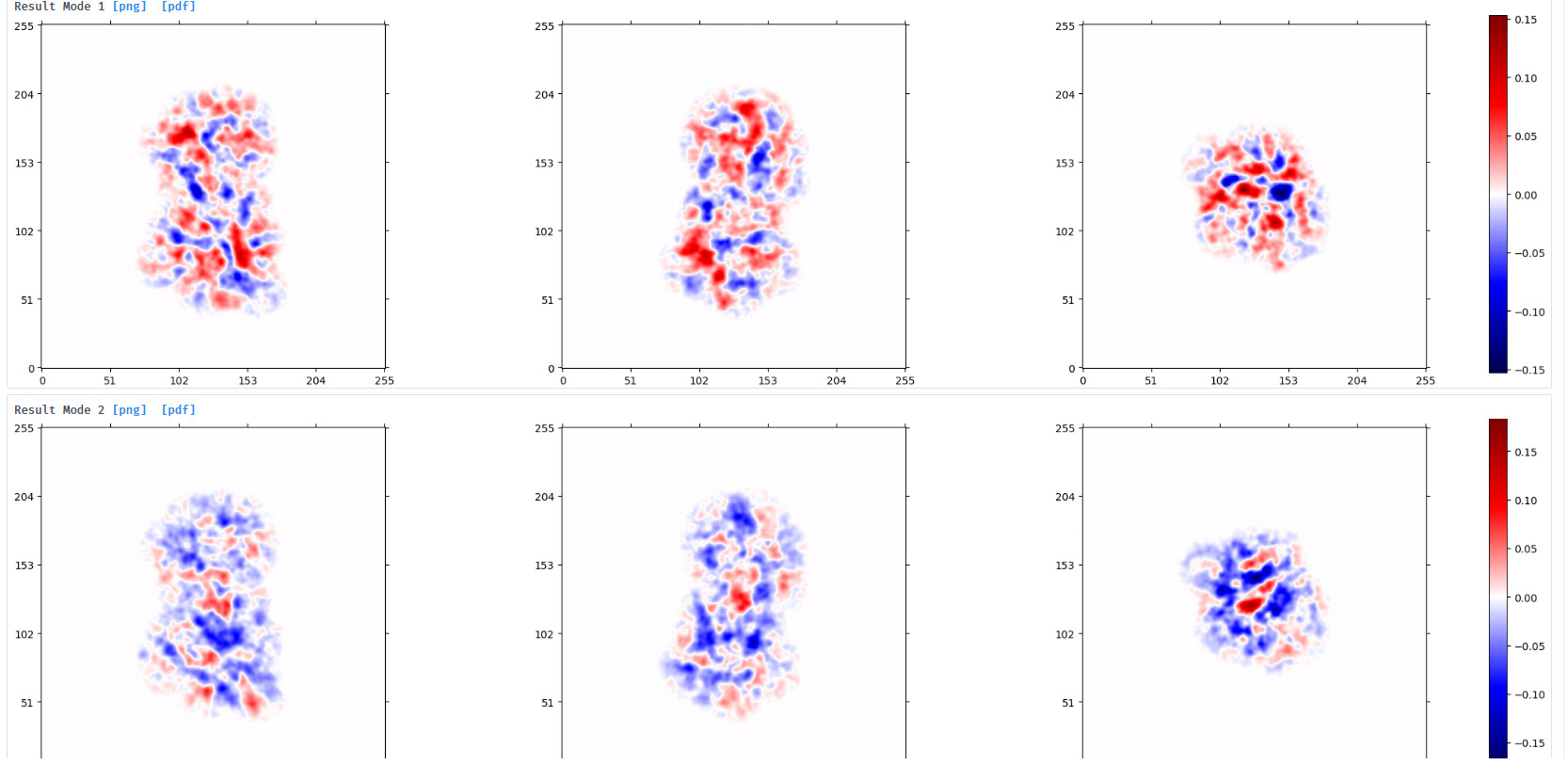
Output from 3DVA:
component 0 ‘simple’ display movie (purple) and component 2 ‘simple’ display movie (green):


Output from clustering with 5 clusters:
UPDATE:
I have since performed NU-refinement on the 5 clusters shown above and obtained maps of around 4-5A for each (there’s still likely heterogeneity due to glycosylation and each cluster is only of around 20-30k particles which would decrease SNR and limit resolution).
When I morph between maps in Chimera I observe the motions shown by 3DVA although a bit less pronounced.
Is this an appropriate way of analyzing my data? Can I now build models into these maps (consensus plus 5 clusters)? The consensus has 120k particles and better features in some regions likely due to higher SNR.
The motions may be less pronounced following separation into clusters because the cluster output does do not give ideal separation for the individual modes of variability, as it is working in 3D coordinate space rather than 1D. This will also reduce the number of particles in each reconstruction.
Since you know your data are good to at least 5A, you could try modelling/refining into the ‘simple’ output maps filtered to that resolution. I would just avoid reporting GSFSC resolution for those frames, but rather indicate that a manually-set resolution cuttof was used.
Thanks for the reply @Ablakely!
I never thought that it would be acceptable to build models into the ‘simple’ mode maps but appreciate the suggestion. In that case, would you recommend using the extremes of each mode for model building? Would such maps only be used for display in a publication and not deposited?
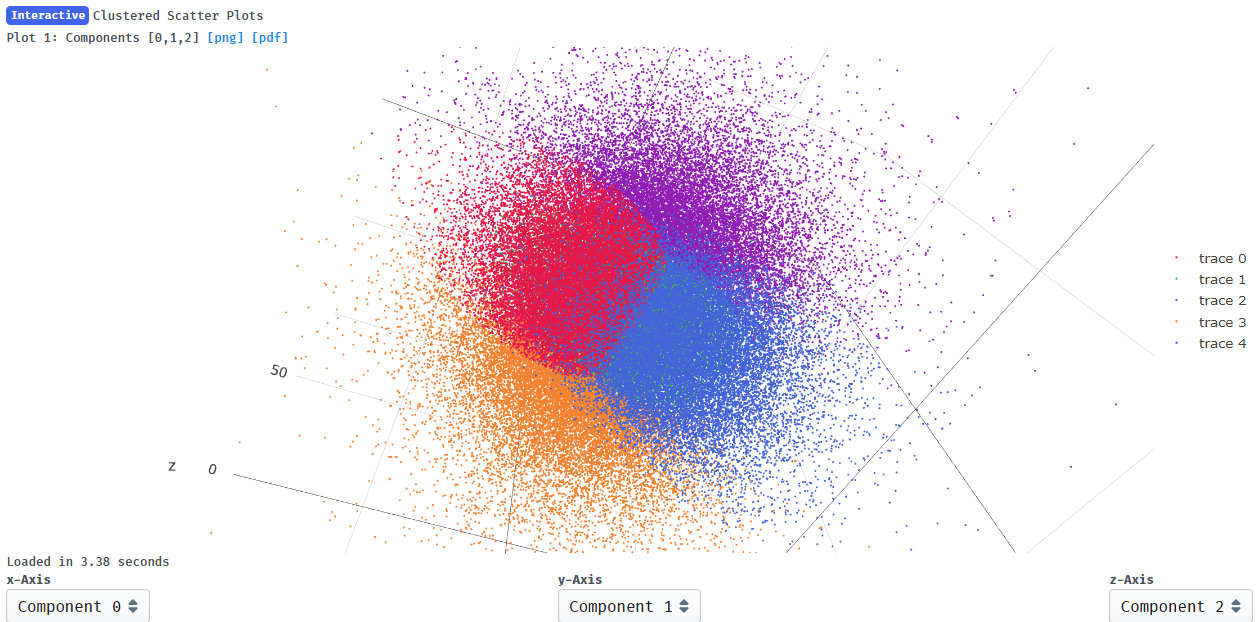
I have since repeated the consensus refine with a tighter mask (it had to be very wide in early jobs to avoid FSC artifacts) and saw slight improvements in resolution so thought that 3DVA should also be repeated using the improved alignments (and a dilated mask for 3DVA). This helped indeed and the motions are more pronounced than before. I again performed clustering and refined those particle stacks. Although the 5 clusters are not distinct in the interactive display, I see a range of helix positions and overall resolution of around 4.5-5A for each. Maybe I should then just build models into the clusters instead?
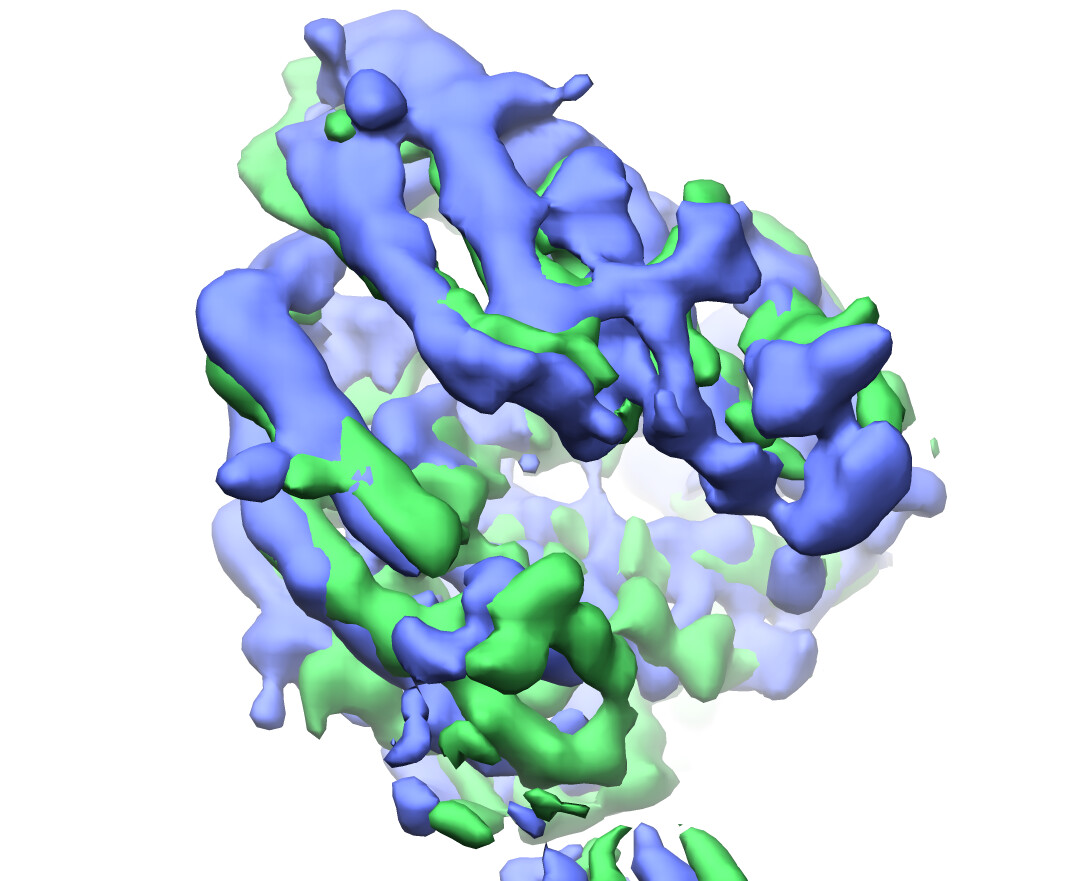
Frame0.mrc and frame19.mrc from component 0 respectively compare very well to the refined clusters 4 and 2.
grey mesh=frame 0, blue surface=cluster4:
yellow mesh=frame 19, green surface=cluster2:
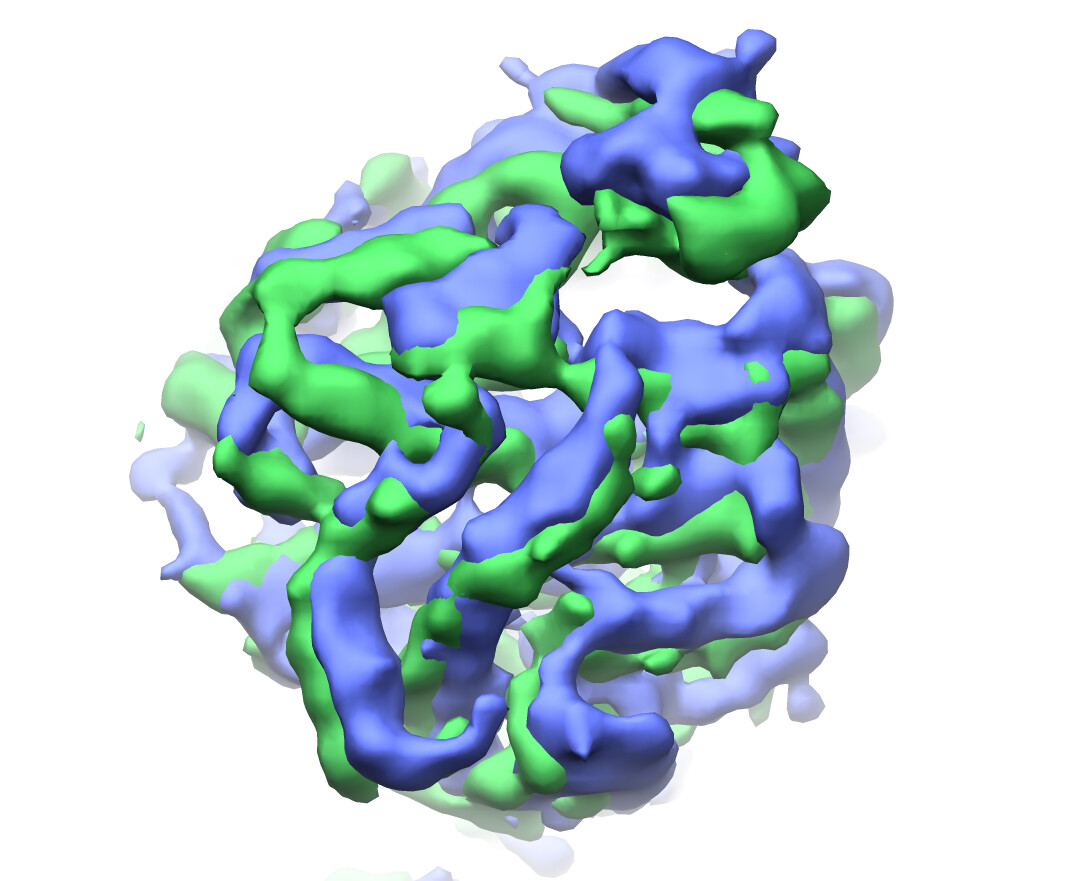
refined cluster 2 (green) vs cluster 4 (blue):
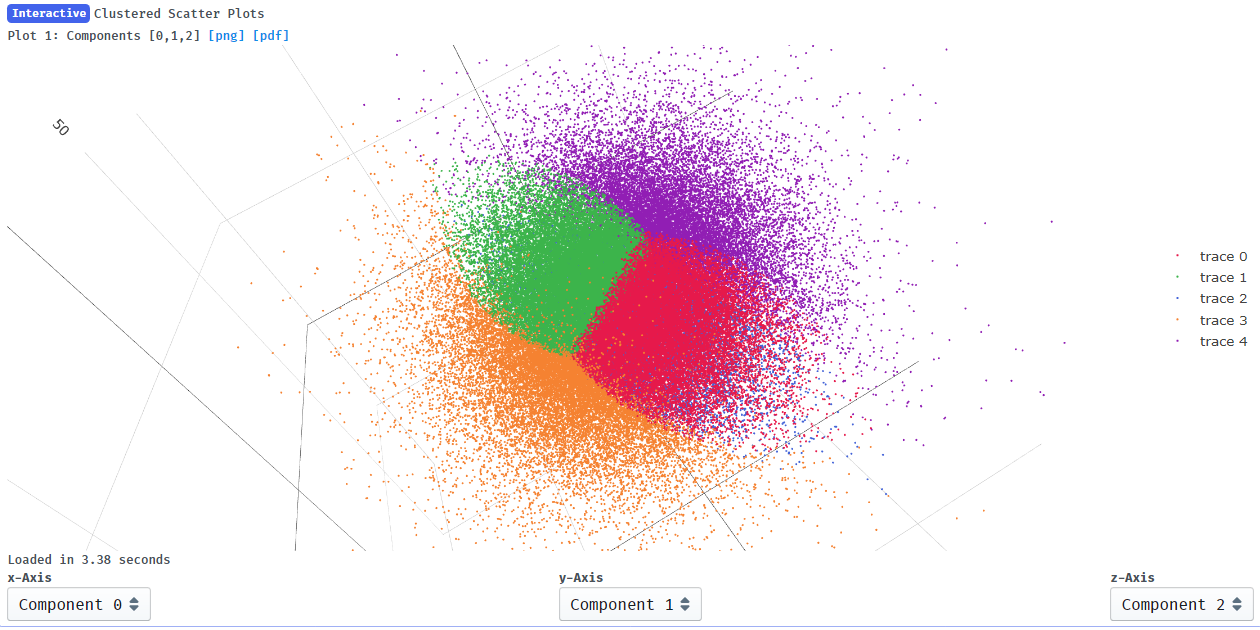
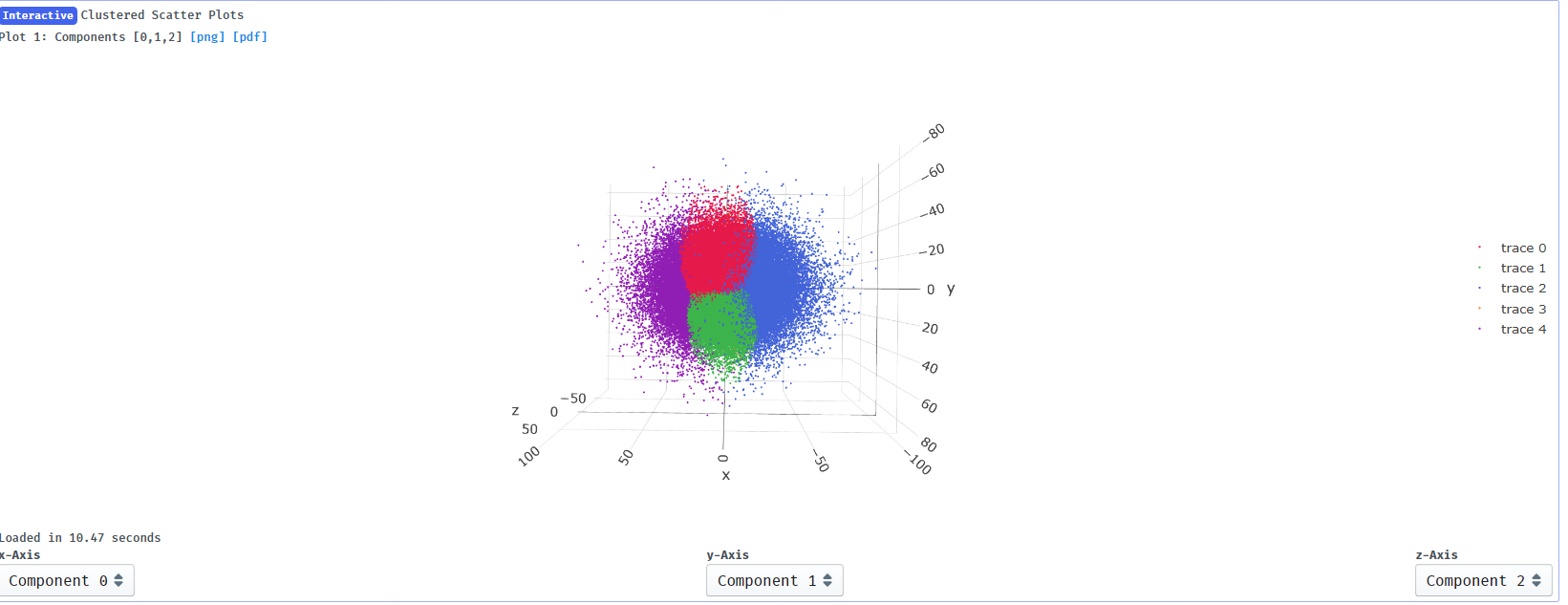
I suppose it makes sense that clusters 2 and 4 show the greatest change in component 0 since they are at the extremes of x on the clustering plot:
You could build into the cluster output as well. If you are limited by particle numbers then it isn’t ideal though, as you can see in your last image cluster 0, 1 and 3 are essentially the same as far as component 0 is concerned, and those particles are split into 3 clusters rather than 1. Since the variability modes are uncorrelated I can’t think of a good reason why one would want to cluster in 3 dimensions, but perhaps I am thinking about this incorrectly.
On the other hand, if you are happy with the resolution/ particle number per cluster, having real refinements done on the clusters is an advantage for reporting or depositing the structures when you publish (without having to do your own python scripting to get 1-dimensional clusters).
Having more particles didn’t give me much higher resolution (the consensus was at 4.1A global resolution and visually only slightly better than the 4-5A clusters) . Probably because more particles equaled more heterogeneity and thus again, decreased resolution. Looks like the small size, high glycosylation and flexibility ultimately limits the achievable resolution in this case. The defocus range used in data collection was -1.8um to -3.0um so perhaps that is also limiting? At present my box size is 256pix for a particle of ~180A at 1.06apix so maybe an increased box size could help with resolution?
I can try to postprocess these maps by local resolution filtering and then build into the consensus map (where some parts of the active site core are better resolved) and the clusters (to describe the motions).
Hi!
Given how small your protein is, I don’t think your defocus range was bad (I could be wrong though).
Increasing your box size might yield better results; given that your protein is ~170 pixels (with your pixel size) in diameter, you could try ~340 as a box size in order to grab all the CTF delocalization. I don’t have experience with proteins that small, but using 2x your particle diameter as a box size is generally not a bad thing to do.
Best,
Armando
I would definitely try a larger box size and see if it helps, given you collected at relatively high defocus and currently have a pretty tight box.
Hi @ArmandoPach and @olibclarke,
I have since tried to increase the box size but unfortunately got worse results as explained in this post Why does smaller box size give better resolution? - CryoSPARC Parameters - cryoSPARC Discuss.
I also thought the box was a bit tight but the limit seems to be 256pix for the monomer and 360pix for the dimer. Thanks for the advice though!
2 Likes
Hi everyone,
I am resurrecting this old post of mine.
After applying more cryosparc tricks the past few months, I obtained 3.6-4A resolution maps for the different parts of my proteins and have built and refined atomic models with good validation metrics.
The dynamic motions that were revealed by 3DVA (movie earlier in the thread) are interesting and I want to model the different 3DVA components using my high res models. Can someone please give me some advice on how to go about this? I understand that the 3DVA movie frames from the ‘simple’ output are not real reconstructions so I don’t know what is ‘allowed’ i.t.o modeling into them and interpretation of those models. I have decided to use the simple mode and not cluster mode output as the particle numbers were too low for refinement after clustering.
@Ablakely, you mentioned that I can build into these frames if I avoid reporting the GSFSC resolution and filter the frames to something like 5A. Would you recommend building CA traces only or could I use the 3.7A full atomic models that I have refined? If the latter, I suppose I can use distance and torsion restraints on the current models in Isolde and just let them relax into the different movie frames’ maps? I don’t know if I can deposit such models but want to identify where these motions are occurring. I haven’t found a literature example of models into 3DVA frames, only reports showing the movies, so please point me in the right direction if I missed any literature.
1 Like
Hi @lizellelubbe, you can also use “intermediates” mode, which does calculate reconstructions of particle sets along the axis of the relevant 3D-VA component - maybe this would help?
Hi @olibclarke
I also calculated the intermediates but remember them being rather noisy (the transitions weren’t smooth and the maps had sharp edges all over). Don’t know if I set a parameter wrong or something.
hmm they should be fine, I usually use intermediates mode with no issues. The only thing to tweak might be the min/max range percentile and the number of frames - if you don’t have many particles, then the default of 3% might leave the start/end frames a bit noisy (and the default number of frames - I think 20 - will also be too high, leaving you with sets in each bin that are too small for high resolution reconstruction). I usually do 5 frames with no overlap (window 0), and that works pretty well
1 Like
Okay I will try again. My frames were set to the default of 20 and I didn’t adjust the percentile range. Both particle sets are 110k with the one then also having symmetry expansion in C2 before 3dva. I will play around with the number of frames, range percentile and window. I somehow didn’t realize that the particles were actually split by this algorithm. Just checked the tutorial again and the parameters make more sense now! Thanks for sharing what works for you