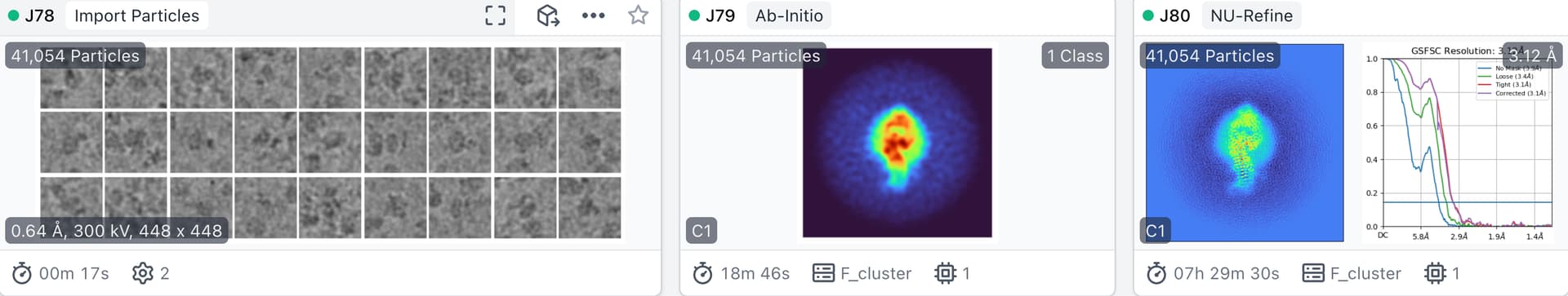
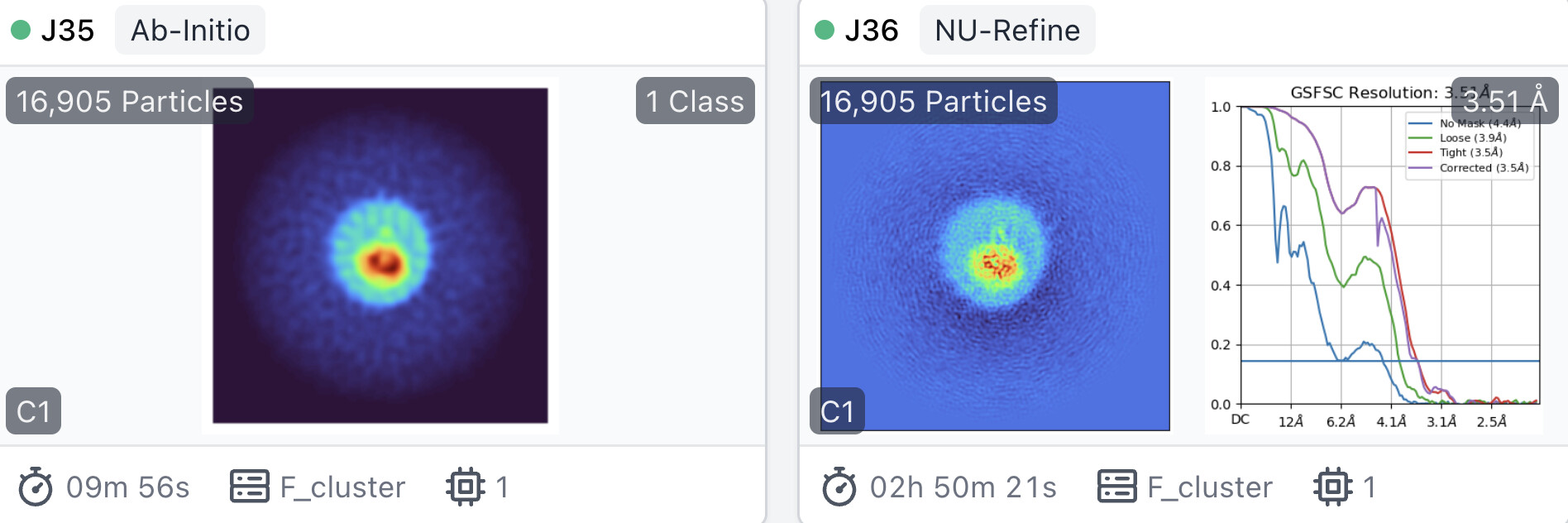
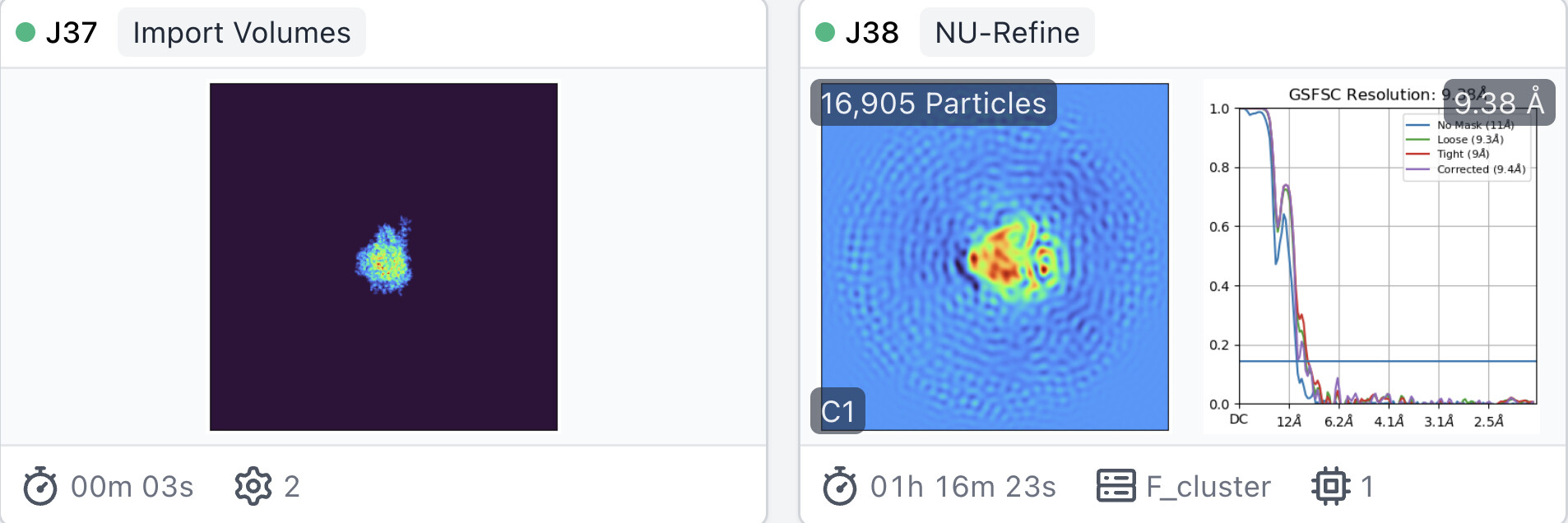
I tried two ways of model initialization of non-uniform refinement:
ab initio reconstruction with C1 and default parameters(Number of Ab-Initio classes is also 1).
atomic model generated by pdb (In Chimera, molmap and resample to corresponding size).
BTW, the parameter ‘initial lowpass resolution (A)’ in non-uniform refinement is 30A by default.
Surprisingly, the resolution of lowpassed atomic model initilaized NU-Refine is much worse than that of lowpassed ab-initio reconstruction initilaized NU-Refine.
Lowpassed atomic initialization is more accurate and should produce a better, at least similar map, not worse. I wonder if this phenomenon is related to non-uniform refinement itself.
I’ve seen this every time. same for Het refine where it will put virtually all particles to the classes generated from data and none to the imported model. I believe it’s a grey-scale issue leading to a funky noise model. There are some parameters affecting this that you can change, but in general I try to use only data-derived references for this and other reasons. BTW same problem in template picking.
One thing to note is that the atomic model doesn’t include density in the micelle, whereas the ab-initio model from data has nonzero density where the micelle is. For the purposes of initializing a refinement, the micelle density is definitely important as it helps align the particles at low-resolutions. This is likely explaining the failure you’re observing in the atomic model initialized refinements.
For future reference, it may be possible to initialize refinements from plain unmodified atomic models for densities that don’t have large regions of disorder – e.g. for highly symmetric particles, viruses, apoferritin, T20S, etc., but membrane proteins probably wouldn’t work this way from unmodified atomic models.