Hi There,
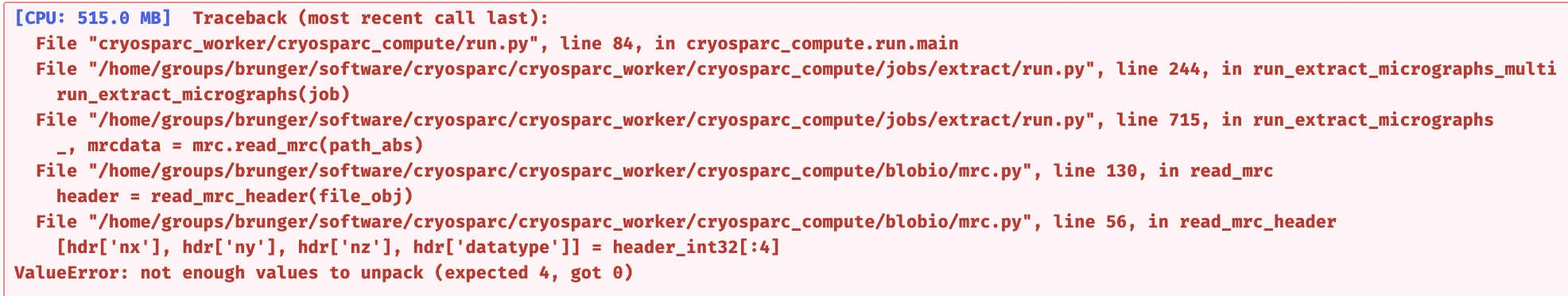
I’m processing some SPA data with gold beads, so I subtracted the beads signal from motion-corrected micrographs, then do CTF, template picking, but reported an error when doing Extract from Micrographs as below:
Hi There,
I’m processing some SPA data with gold beads, so I subtracted the beads signal from motion-corrected micrographs, then do CTF, template picking, but reported an error when doing Extract from Micrographs as below:
Hi @CleoShen it looks like cryoSPARC is having trouble reading the associated background micrograph for each exposure. The file may be empty or corrupted. What job or program did you use to do the motion correction?
If you used a cryoSPARC job, can you show me a directory listing of the the motion correction results with file sizes? You can get it from the command line like this (substitute the project path for you system and job number of the motion correction job):
cd /path/to/P3/J42/motioncorrected
ls -lh | head -n 50
Then can you also send me the background data file for input micrographs for the job? You can get them from the cryoSPARC interface in the outputs to the motion correction job, as per the screenshot:
The file may be a few hundred MB depending on how many micrographs you have, so you may have to use a file sharing service like Dropbox.
If you didn’t use a cryoSPARC-provided motion correction method (such as Motioncor2) I’d suggest also trying the Patch Motion correction job and see if that makes a difference.
Hi nfrasser,
Thank you for your information. I fixed it. I did some subtracting gold beads processing on the background files. I tried again by using unprocessed background files, the job works now.
Hi nfrasser,
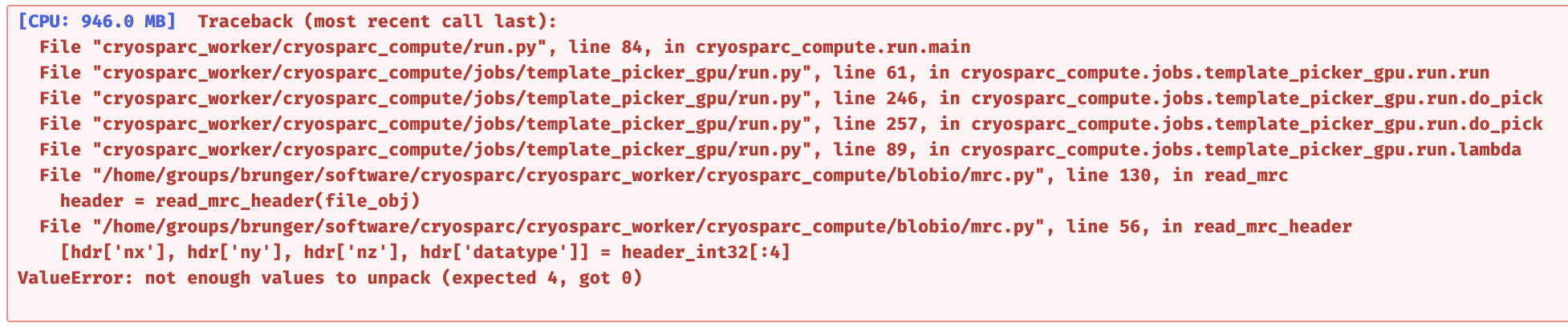
I had a similar error when doing the Template Picker job:
The weird thing is I tested the step 1 to 4 in a dataset of ~1.5k micrographs, it goes well. But for the dataset of ~3k micrographs, it has this error when doing the step 4. What can I do next?
This again looks like one or some of the micrographs ended up corrupted, but this time it’s the full dose-weighed micrograph (some *_aligned_doseweighted.mrc file).
Can you again try navigating to the motioncorrected directory and run ls -lh to see if any micrographs have file size 0, or differing significantly from the others? You would then have to re-create these files to get the template picker to work (e.g., re-run the Motion Correction job)
Can I just trash these size 0 files, as I don’t know how to re-create those files?
Unfortunately right now the Template Picker isn’t set up to skip missing files, so the whole job will fail if any exposure file is missing. You may be able to take them out if you first run the exposures through a “Manually Curate Exposures” job and reject the exposures with missing previews. Try that out and let me know how that goes, if it doesn’t work we’ll try to find an alternate solution.
Hi nfrasser
One naive question: how to quickly find the individual exposure without preview? I’ve created a Manually Curate Exposures.
FYI. I’ve tried to manually remove all the zero size .mrc files, then do Template Picker, yes, doesn’t work.
@CleoShen unfortunately there’s no quick way to pick out exposures without preview from the Curation job. You have to manually click through the list in interactive mode and see if the previews load, then manually accept or reject those. Alternatively, pick out the zero-size files from your file system and search for those in the interactive list.
I tired the way you said, but as I have about 16K micrographs in total and there is no quick way to locate the zero-size files in the interactive list instead of open each micrograph one by one. This solution is so time consuming. If I have other choices, like delete all the zero-size .mrc files and correlated .npy files, then do CTF again?
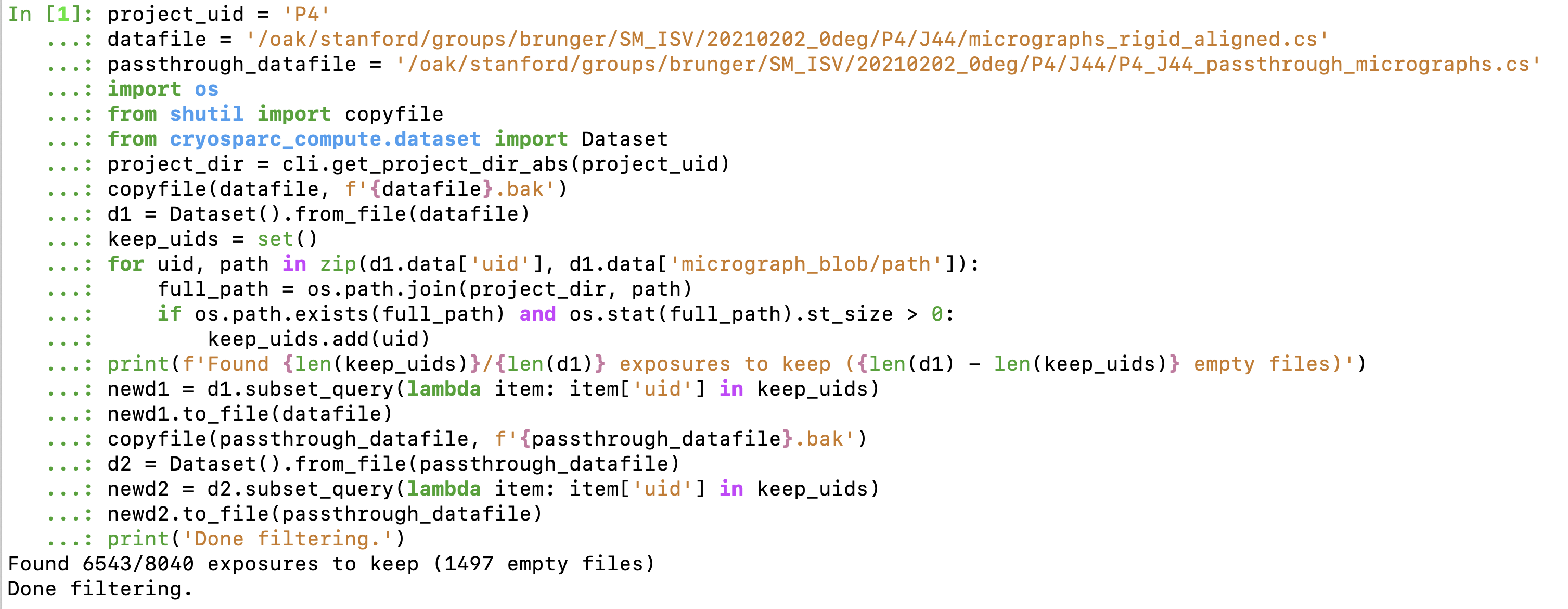
Okay, I’ve put together a script you can run to modify the results of the motion correction job and filter-out zero-sized files, here’s how to run it:
project_uid = '<PROJECT UID HERE>'
datafile = '<OUTPUT DATA FILEPATH HERE>'
passthrough_datafile = '<PASSTHROUGH OUTPUT DATAFILE HERE>'
import os
from shutil import copyfile
from cryosparc_compute.dataset import Dataset
project_dir = cli.get_project_dir_abs(project_uid)
copyfile(datafile, f'{datafile}.bak')
d1 = Dataset().from_file(datafile)
keep_uids = set()
for uid, path in zip(d1.data['uid'], d1.data['micrograph_blob/path']):
full_path = os.path.join(project_dir, path)
if os.path.exists(full_path) and os.stat(full_path).st_size > 0:
keep_uids.add(uid)
print(f'Found {len(keep_uids)}/{len(d1)} exposures to keep ({len(d1) - len(keep_uids)} empty files)')
newd1 = d1.subset_query(lambda item: item['uid'] in keep_uids)
newd1.to_file(datafile)
copyfile(passthrough_datafile, f'{passthrough_datafile}.bak')
d2 = Dataset().from_file(passthrough_datafile)
newd2 = d2.subset_query(lambda item: item['uid'] in keep_uids)
newd2.to_file(passthrough_datafile)
print('Done filtering.')
<PROJECT UID HERE> with the ID of the project you are in, e.g., P3
<OUTPUT DATA FILEPATH HERE> with the path to the micrograph_blob output of your motion correction job. Copy this value from the job’s “Outputs” tab, as indicated:<PASSTHROUGH OUTPUT DATAFILE HERE> with the path from one of the outputs marked as “passthrough”, e.g., the mscope_params outputcryosparcm icli
Let me know if you have any trouble with that.
Hmm, maybe some of micrographs aren’t zero sized? Can you check the the motioncorrected folder again and see if there are any files with a very small size? e.g., less than one 100 bytes?
If you find any try re-running the same script but replace this line:
if os.path.exists(full_path) and os.stat(full_path).st_size > 0:
with this:
if os.path.exists(full_path) and os.stat(full_path).st_size > 100:
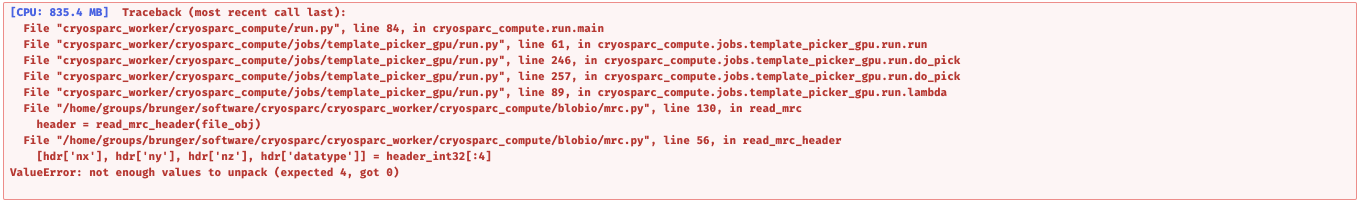
Oh, I might make a mistake. If I should trash all the 0 size files before running the script you shared with me? I left the 0 size files for the last error try shown above.
This wouldn’t be related, the code I sent does not affect the contents of the database. This error can happen if cryoSPARC didn’t restart properly and there are some artefacts left over from the previous processes. Can you send me the output of the following commands?
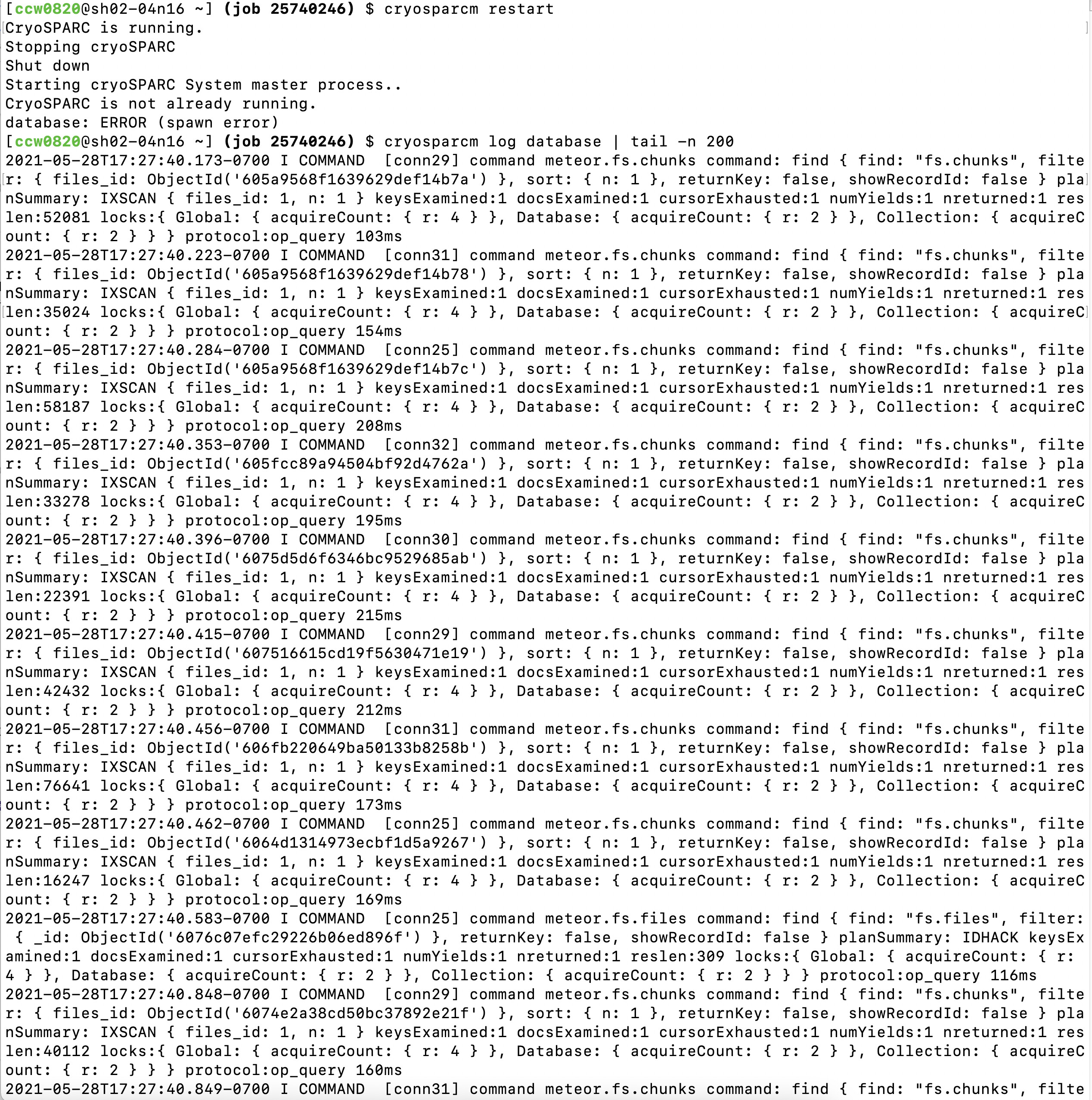
cryosparcm restart
cryosparcm log database | tail -n 200
I’d also suggest restarting the machine on which cryoSPARC is running.
Hi nfrasser,
Thank you for your reply, here pasted the log below. BTW, I’ve tired to restart my iMac and terminal, but can’t restart the computer cluster, FYI.
It looks like there’s something else running at the network port that cryoSPARC should be running on and this is preventing the database from starting up.
Can you confirm what your base port is? You should see it inside cryosparc_master/config.sh - look for the line that starts with export CRYOSPARC_BASE_PORT= - the proceeding number will be the base port.
Then, only if the base port is 39006, follow the instructions I posted here: Regarding installation of version 3.2
But instead of starting the lsof steps at 39000, start at 39006. e.g.,:
lsof -i :39006
lsof -i :39007
lsof -i :39008
lsof -i :39009
lsof -i :39010
lsof -i :39011
lsof -i :39012
lsof -i :39013
You also don’t have to run any of the commands that begin with cd and you should use cryosparcm instead wherever you see ./bin/cryosparcm.
After you’ve killed the processes bound to any port, run those lsof commands to ensure the processes are not running.
Then start cryoSPARC with cryosparcm start
If the base port is config.sh something other than 39006, let me know here what it is and I’ll follow up with additional instructions.