I’m working on cryo-EM reconstruction of a membrane protein complex in CryoSPARC, and I’m observing two distinct populations in my 3D refinements. One population has a specific structural feature in the expected orientation, while the other appears rotated ~90° relative to the first.
I’m relatively new to the field and have been trying different approaches to improve resolution. Here’s what I’ve tested so far:
Template Picking (Curated Exposures)
Box size 600 → The 90° rotated population appears.
Box size 700 → The expected orientation is observed.
Template Picking (Whole Exposures)
Box size 600 → The 90° rotated population appears.
Box size 700 → The expected orientation is observed.
Topaz Training & Extraction (Box 700)
Ab-initio Classification Results:
90° rotated population → 120K particles.
Expected orientation → 45K particles.
For all refinements, I start with homogeneous refinement (C1 symmetry), followed by non-uniform (NU) refinement with C2 symmetry.
Has anyone encountered a similar issue? Could this be due to flexibility, or is there a processing step I should revisit? Any suggestions on how to resolve or further analyze this issue would be greatly appreciated!
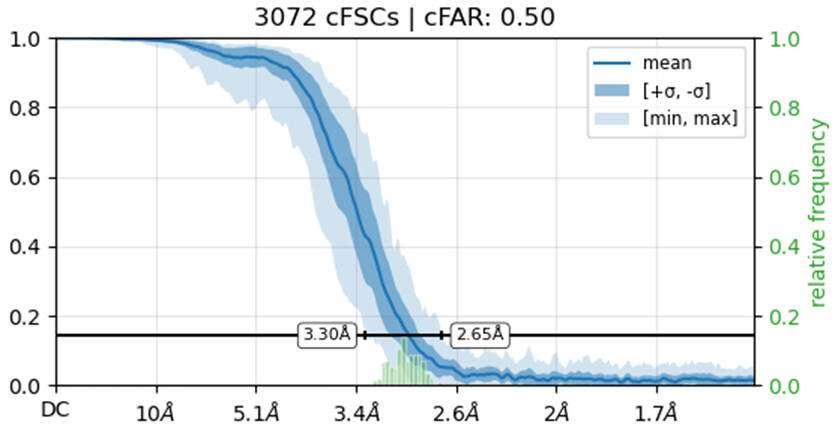
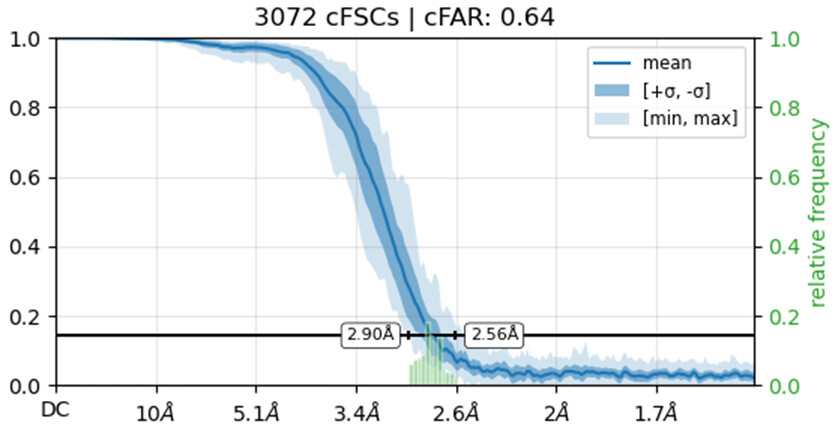
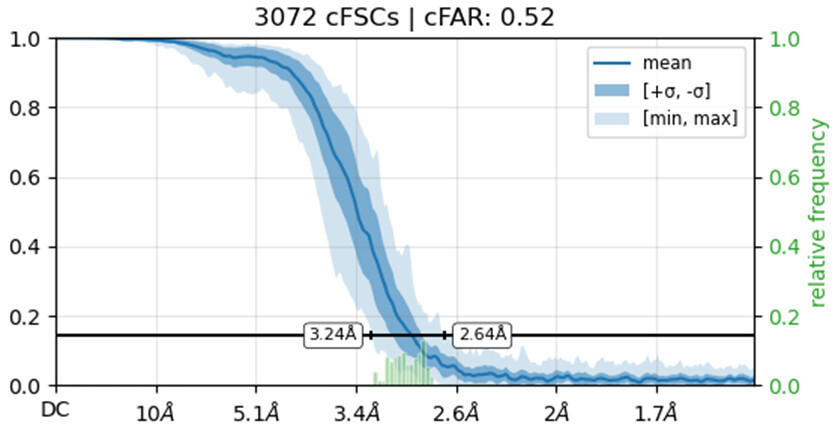
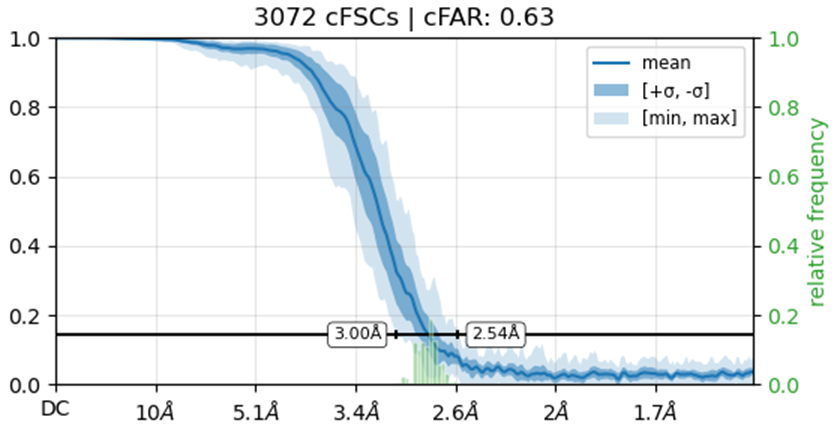
Would you be able to post some images of your volumes that illustrate this issue including both the C1 and C2 refinements? Additionally, can you post the cFAR and FSC plots for the refinements as well.
sorry for the late reply.
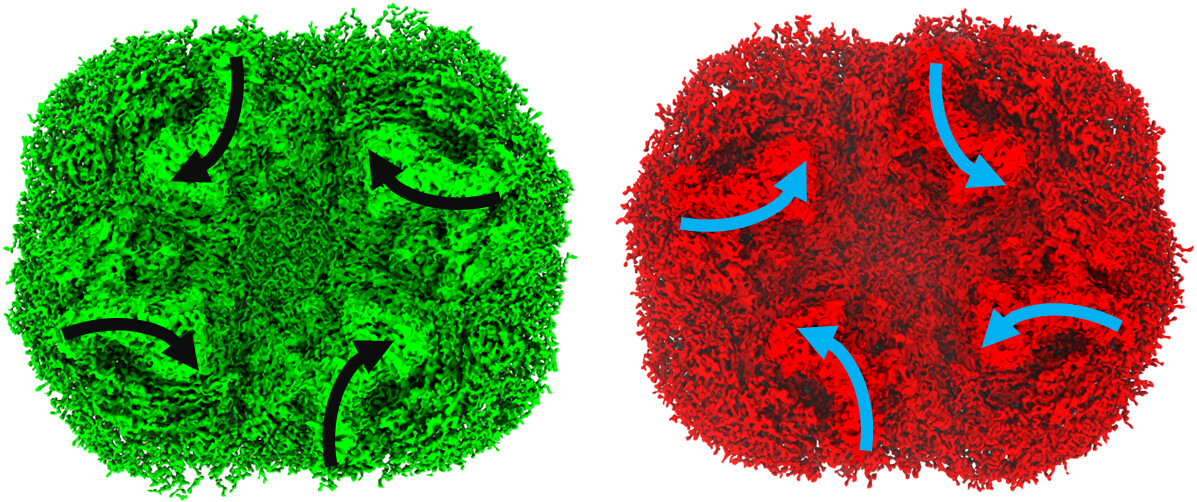
Here is an image for the right orientation (green) and the unexpected orienation (red). C1 and C2 looks same for each orientation. both of them are from the same particles.
The obvious change is in the cytoplasmic side of the protein. the rest of the density looks normal
Have you checked these are not just the correct and inverted hand solution? Because one looks very much like the mirror of the other. Ab initio has equal chance to converge on the correct vs inverted hand solution, so this would be expected
I’m not entirely sure how to check if the solution represents an inverted hand. What I did was run an ab initio reconstruction and obtained two distinct density maps. I then used all particles from both populations to perform separate refinements, using the two ab initio maps as reference volumes. Interestingly, the cFAR appears slightly better (or same) when refining against the orientation that seems unexpected.
To clarify, this protein is Photosystem I, which has a cytoplasmic domain made up of three subunits. From the density maps, it looks like the major difference between the two reconstructions is localized to this domain. Visually, the maps appear to be mirror images of each other, particularly in that region, but I’m not sure how to formally confirm whether it’s truly an inverted hand.
That sounds very much like an inverted hand situation.
What happens if you take the “unexpected” orientation map, invert it using volume flip in ChimeraX (or vop zflip in Chimera) and align it to your other map? If it is an issue of the hand being inverted, they should then align well.
Agree with Oli that you’ve got two different handedness there.
Confirming is easy - try fitting PsaCDE and see if the alpha-helix of PsaD fits or is backwards. Or, as explained, zflip one in Chimera and compare.
Also make sure you haven’t got any duplicate particles because those FSC curves do not reach zero.
The simplest solution is take the correct handedness map (confirm by fitting a PDB) and refine the whole stack using that as a reference with homogeneous/NU refine. Then 3D classify to remove junk or tease out any subset which may have something interesting bound (e.g. ferredoxin).
Tetramer PSI cores? Interesting… red alga?
Handedness is always a coin toss. Theoretically 50/50, but with initial models I get inverted handedness 90% of the time.