Hello everyone
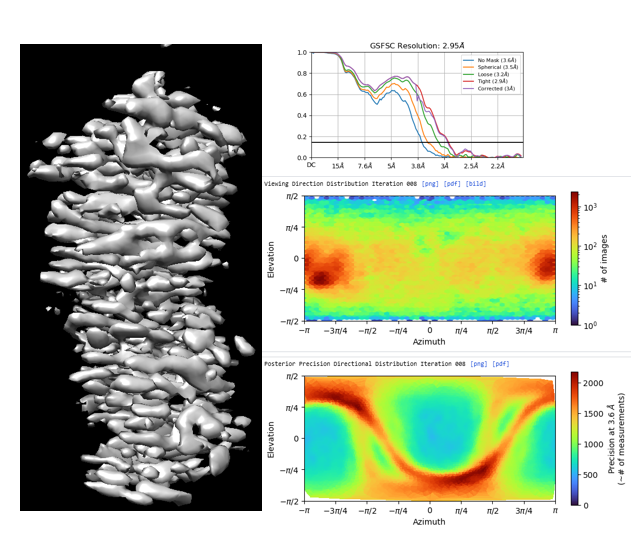
I am working with 150 kDa protein, at the level of 2d classification and ab into constructions, the processing is normal. once the map reaches high resolution, I observe this stretchy pattern in the map. I tried to rebalance the 2D classes in addition to iterative cycles of 3D variability analysis and 3D classifications, heterogenous and NU refinement but failed to eliminate this problem.
Welcome to the forum. ![]()
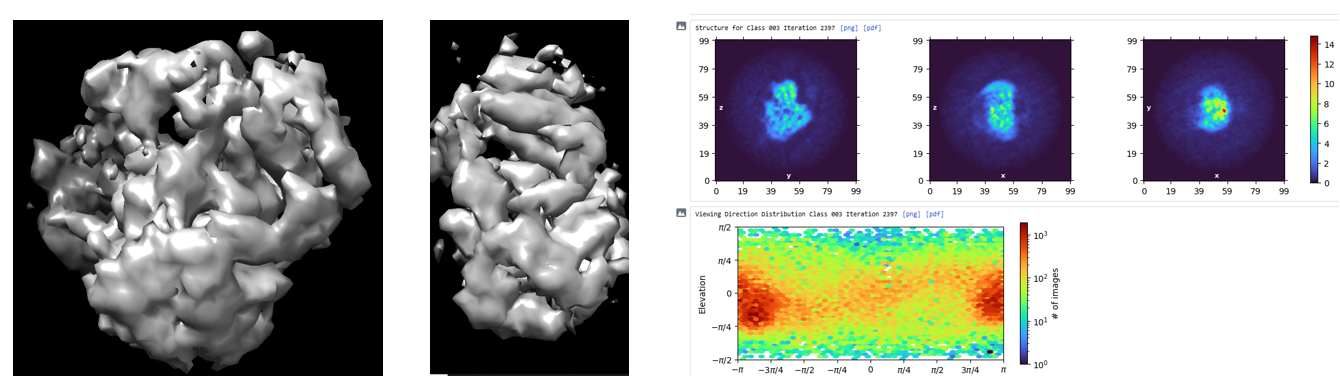
What does the ab initio look like in 3D?
The direction distribution shows a strong orientation preference, although the map certainly looks worse than I would expect given that you do have some distribution outside of the preferred direction…
Try to select fewer particles of the dominant view in 2D. Also try heterogeneous refinement…
Thanks so much for your answer
the initial model has the general shape of the protein with less intensive stretches. I have already filtered the particles by removing dominant views and enriching the rare views but still failed to eliminate the stretches.
Hi Mahmoud,
What does the output of the Orientation Diagnostics jobtype look like (for the refinement in your first post)?
Cheers
Oli
This is one of the most common issues in cryo-EM. Getting more particles of a different view is paramount. If there are a few 2D classes that are unique (represent side views): select all 2D NOT corresponding to dominant view, rerun 2D with batchsize 1000, select all good particles along with 1 or 2 classes of the dominant view and try refinements.
Alternatively, het refine and 3D class can occasionally, and somewhat randomly, select a single great set of particles that sample all views. they also give very high resolution classes that contain dominant view only, so need to learn how to interpret this result and ignore these classifications. for het refine, try a bad (above) volume and several ab initio as inputs, and set lowpass to 10 not default. for 3D class, try several values for expected resolution, maybe 4,5,6,8,12 in distinct jobs.
If still stuck, you could try building an appropriate model in chimera, molmap and resample, import volume, create templates, 2D select rare views, template pick, 2D, 2D select rare views (from your data!) and topaz train using these. And iterate this procedure - topaz can keep finding good particles. Be careful though to use “remove duplicates” frequently as it also duplicates particles constantly.
2 Likes
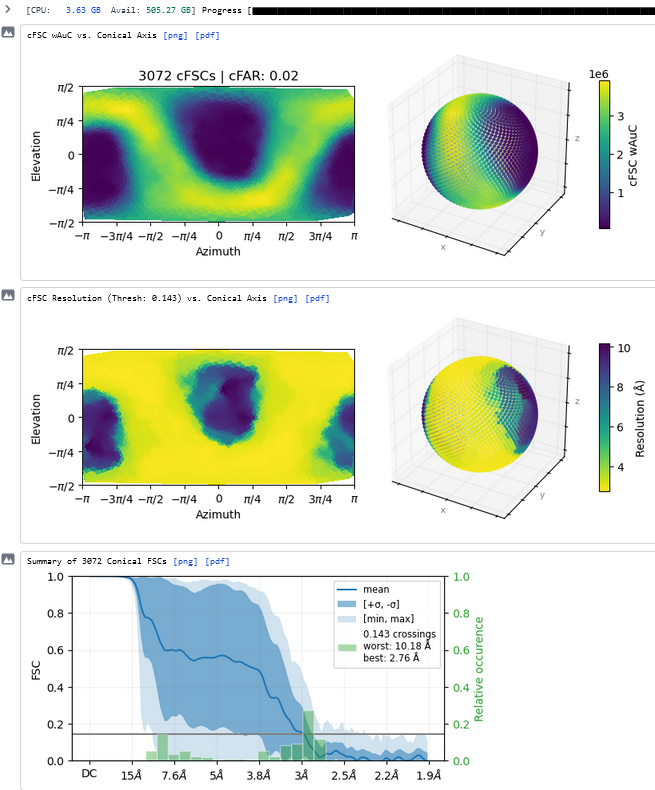
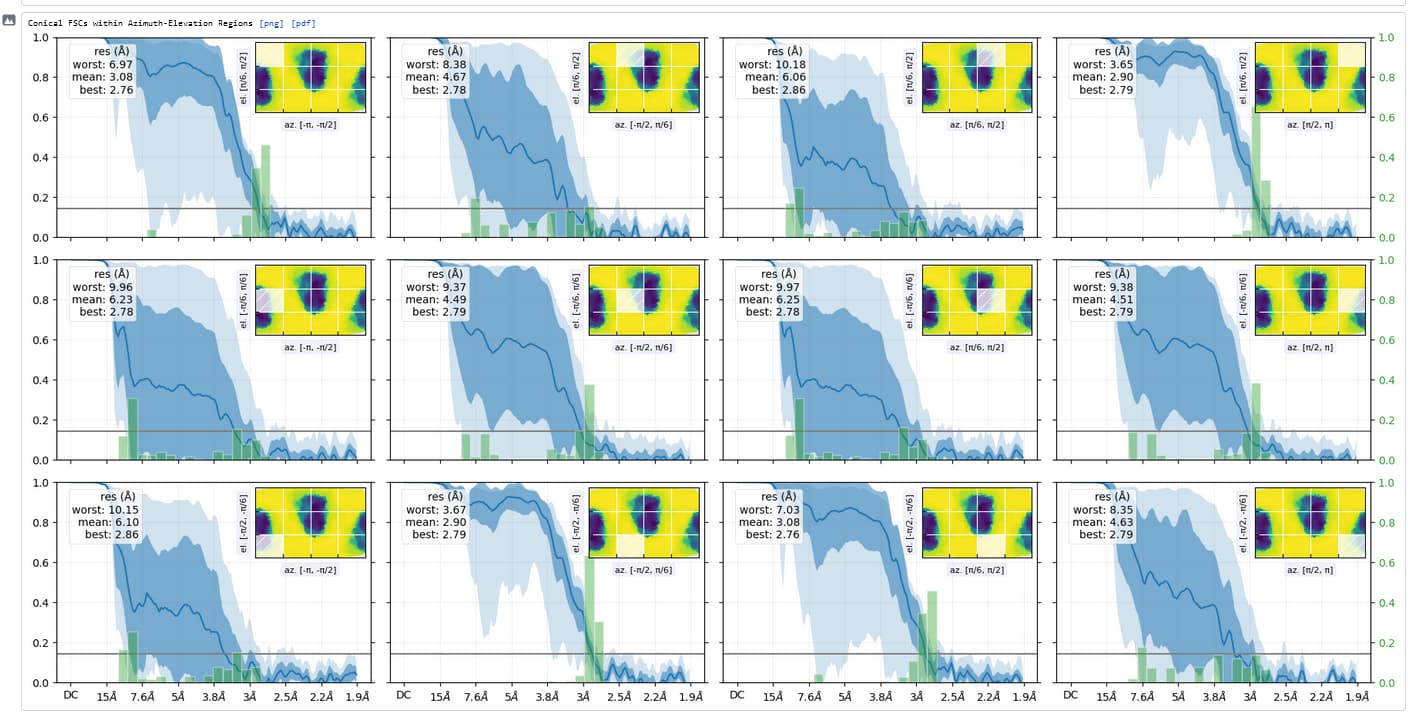
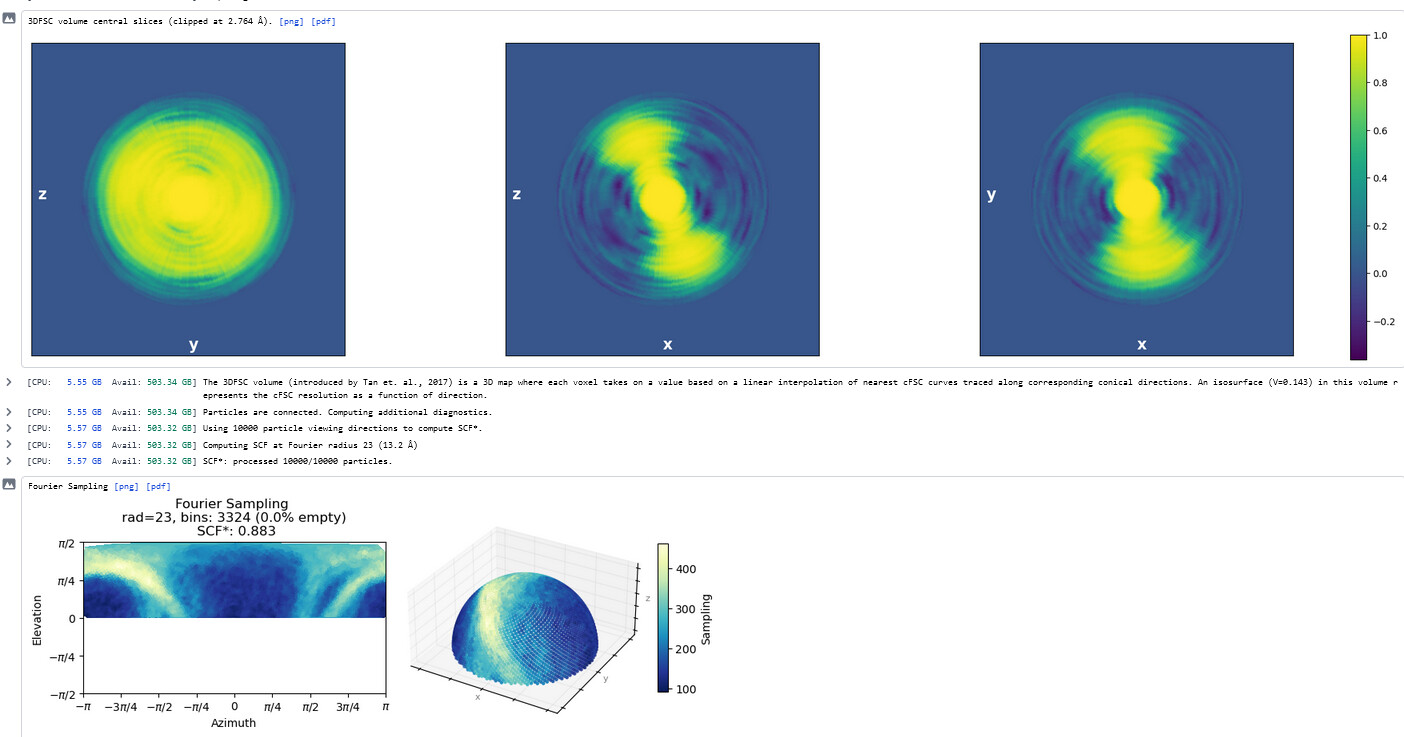
Thanks - this shows the preferred orientation very clearly - 2.8Å in best direction and 11Å in worst!
I would (apart from good suggestions already mentioned to deal with the data you have in hand) suggest screening some high CMC detergent additives to mitigate orientation bias, such as CHAPS or FOM.
Also might be worth collecting a dataset with fixed tilt, to recover some views.
Cheers
Oli
3 Likes