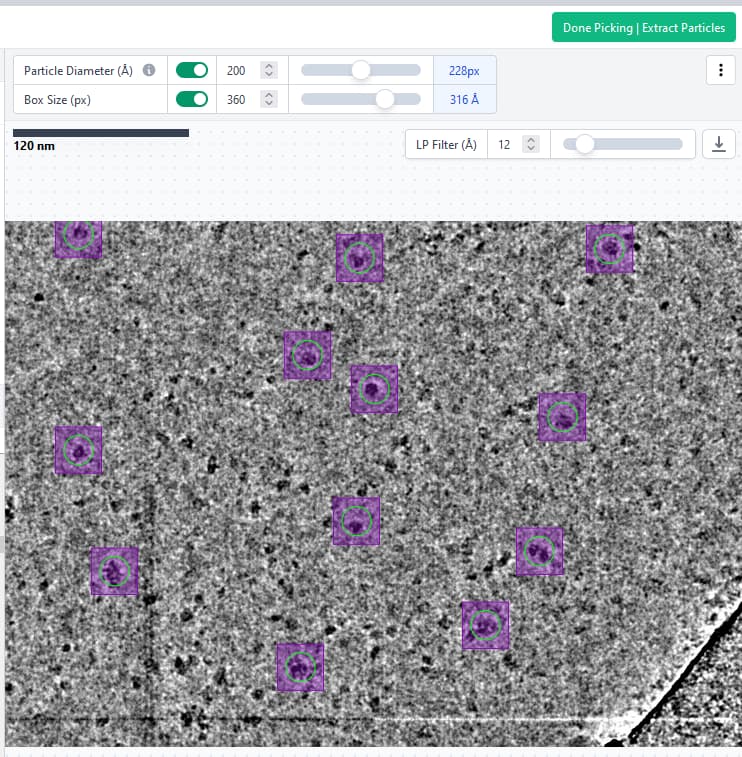
The parameters I used were:
number of 2d classes: 100
Force max over poses/shifts:off
batchsize per class: 200
number of online iterations: 20
full iteration: 1.
Any tips, how I can get reasonable looking class averages?
Thank you
Cheers!
If you have Force/max off, it will take a lot longer to converge - usually 40, sometimes up to 80 iterations. Have you tried with Force/max on (the default) to compare?
Yes that’s right, although there are also other things you could try with force/max on - increasing batch size and increasing the number of final full iterations (to e.g. 10) can help
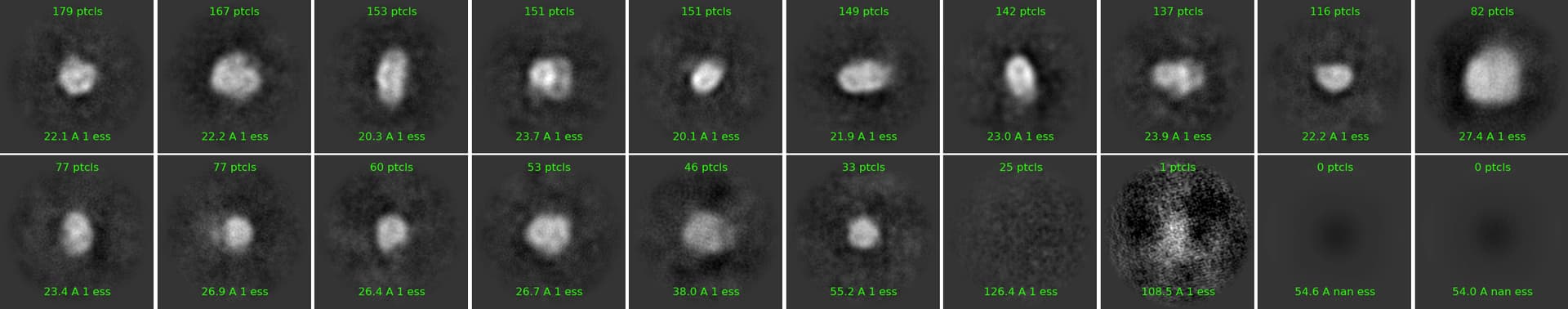
keep in mind 99% of your particles are in the early classes. the spheres are close to nothing. I bet if you take the first classes, use a smaller box, maybe 1000 per batchsize, maybe 40 classes instead of 100, you could pull out something. it’s also possible that what you picked/2D are not protein particles with defined physical shape. take class4 with 367k particles only, run 200 classes of those.
Looks much better! I would pick these and do another round.
Looking at your micrograph, and the first two artefactual classes with the squiggly linear features, I would also double check gain correction, as removing those features may help you.
Thanks!
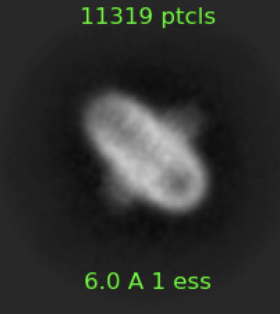
I ran a few rounds of 2D class averaging and ended up with the following classes, which I think look nice. It should be a membrane protein in DDM micelle boudn to a soluble protein.
Was this a his tag purification by any chance? Expressed in E. coli? The top class looks very familiar, it reminds me a lot of complex 4, which is His rich and can co purify on IMAC…
In any case, I would def try a quick round of ab initio and see what you get, and I would also try optimizing your picking (either via template picking, or preferably training a neural network model, e.g. for Topaz).
[omitted], I can see the similarity. I will work on optimizing the picking. According to Masspec our proteins of interests are the most abundant. I hope that is the same in cryo. Thanks for all you tips. I will keep you updated!
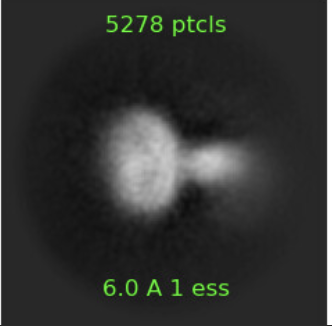
I would also try ab initio with selected classes - e.g. to me these two look more like what you describe (membrane protein with bound soluble protein):