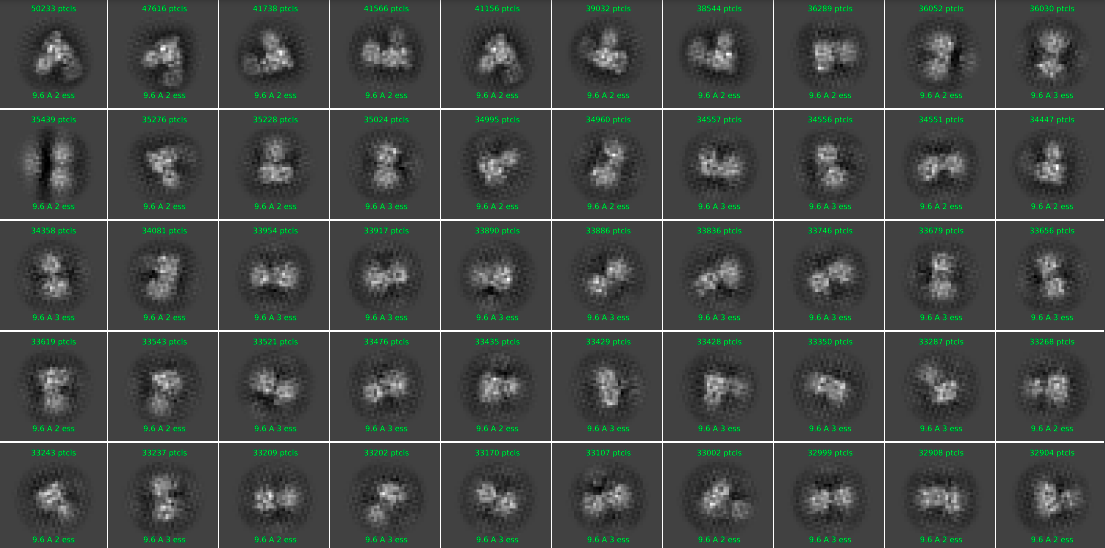
I’m working on a small soluble protein(~80kDa). It gives good 2D classification results (no obvious orientation bias, but it seems that only the front triangular views can be well aligned).
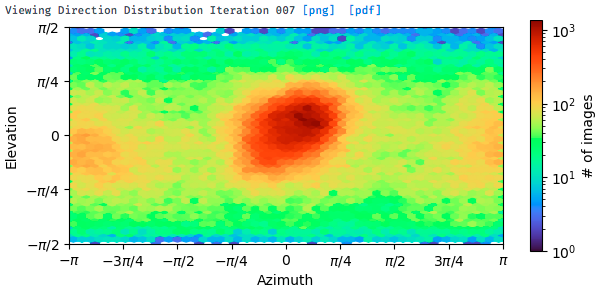
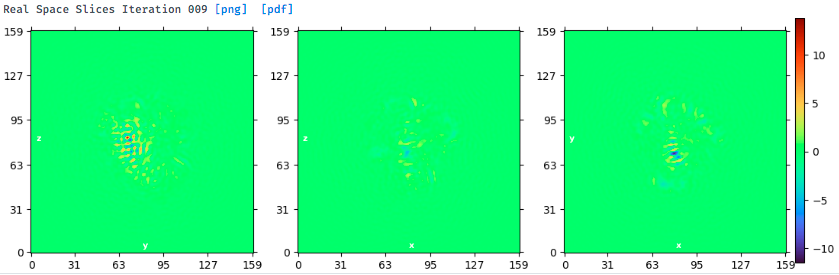
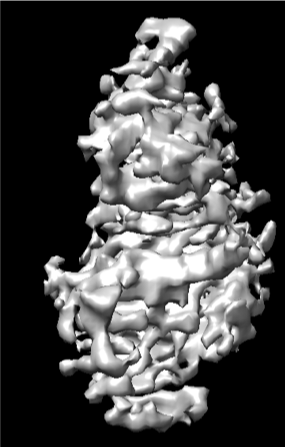
But after heterogeneous refinement, the majority kept particles are those from the front triangular views. The NU-refinement shows severe preferred orientation problem with a higher resolution only for the front view (shown below).
During the processing, I found that the larger “ear” domain is very flexible. Is this the reason for the bad alignment for other views? Is there any suggestions that I can try?
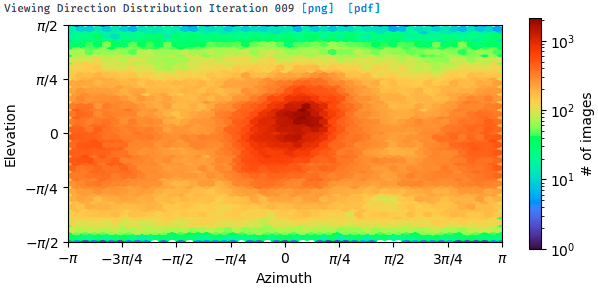
I tried to do seed-facilitated heterogeneous refinement. I got a better viewing direction distribution map, but the signals for certain views are still very weak (as shown below).
Hi ! There is no way you’re getting a sensful ab-initio with the classes you show above; either the ice was too thick, or there is something else going on. How do the classes look like when you switch off Max over poses/shift ìn the 2D class job ?
Hi, Thanks for replying! I just run 2D classification with a subset particles from heterogeneous refinement with Force Max over poses/shift off (680,000 particles, bin1), as shown below.The 2D classes I showed above is downscaled as bin4 (3 million particles).
The 2D classes look already better, probably the binning made them looking worse.
My feeling is that you have in principle a lot of particles to play with, which is necessary when you target this protein size.
My suggestion is the following one:
Split your extract job into 2 or more batches, so that you end up with less than 1 million particles per batch. Do 2D classification for each batch, and increase the number of iterations to 40 with 10 or more final iterations. Remove all classes where you do not see secondary structure elements, or you have a lot of fuzzy density.
Repeat 2D classification till you reach a point where the 2D classes stay more or less the same, and contain only high-quality features.
Use these particles for an ab-initio. Check some other discussions for the parameters that you can change in the ab-initio, for example starting at a higher resolution than default (say, starting at 20 and finishing at 6).
How does you ab-initio looks like ? Does it contain helices, for example ? If so, the refinement will work.
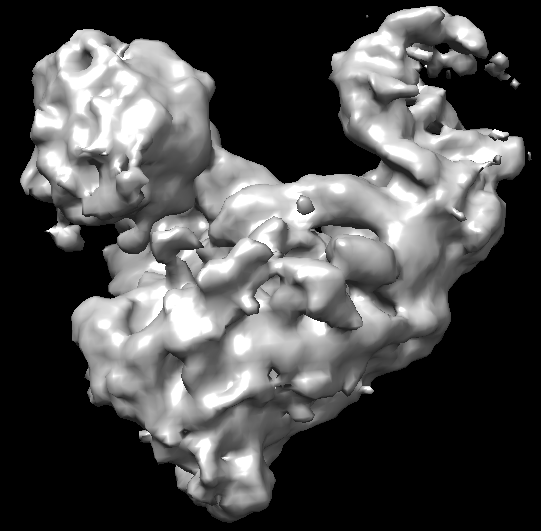
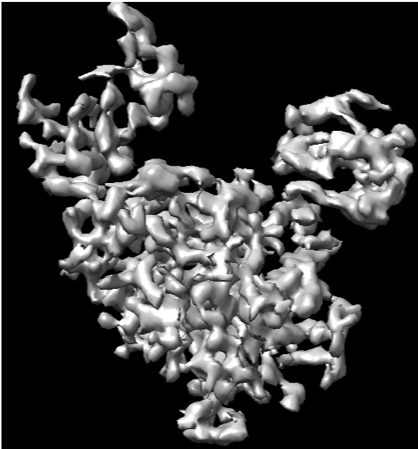
Thanks for your suggestions! I did try lots of ab-initio jobs (with different subset particles and parameters containing set higher maximum and initial resolution), but I don’t think there are secondary structure element in the resulting model. Shown below is a typical ab-initio result.
I’m wondering which strategy would work better if I want to get a better subset particles for ab-initio, removing bad particles with 2D classification or 3D heterogeneous refinement?
I would surely start from extensive cleaning with 2D classification jobs. You probably need 2-300k particles in the final set have a good signal. Once you have your best 2D classes with clear secondary structure elements visible, do the ab-initio. Sometimes 2 classes in the ab-initio help to further get rid of unnecessary particles.
This doesn’t look too bad to me as a an initial model - density looks reasonably isotropic and matches 2Ds. Did you try proceeding to homogeneous or non-uniform refinement?
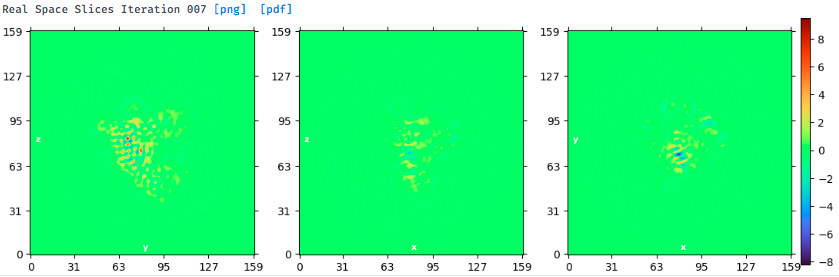
Yes, I tried homogeneous refinement (New) and NU-refinement (both New and Legacy). Usually, the NU-refinement New gives the best results for a given subset. The direction distribution and real space slices of best NU-refinement results are shown above. Below is the map. There is no good secondary structure elements in the map, and the side view shows streaking. It seems hard to get signals from this orientation.
I would say there are signs of secondary structure in the lower view - what looks like a helix in the upper left. Just quite anisotropic (which tracks with the 2Ds). You may be able to improve matters with more classification or better picking, but I would also consider trying some additives to the sample (e.g. high CMC detergents like CHAPSO, beta-OG, FOM) to improve the orientation distribution.
Thanks, Oli! I have a concern about the detergent additives. From the 2D results, it seems the orientation distribution is acceptable. In addition to the front triangular views, there are some top views and side views, but these views cannot be well alignment. People use these additives when the ratio of certain views particles is low. So, I think this may be a different case? I’m wondering would it help by adding detergent additives?
well I don’t know - while there are some side views, to me looking at your 2Ds it looks like the face-on L-shaped views are much more prevalent, and if that is the case then additives may help. Worth a try IMO
It looks like a typical sample with modest preferential orientation to me.
Striving for high resolution you propably have chosen very thin ice, which often exacerbates the situation.
I second Oli, screen a few detergents and see what works.
cheers
from this huge dataset, you could also try 2D classification with/without alignment AFTER the 3D, and manually “remove” lower resolution particles which adopt the same view as higher-resolution counterparts, if they’re in the preferred orientation view. This way you can 1) keep high res data 2) remove excess of the preferred view 3) keep all views which are not overrepresented.
but I echo the others, better data will simplify life and be a fast-track to a good structure.
Hi, thanks for reply! How to run 2D classification without alignment? Is it turning off Force over poses/shifts? I had tried to randomly remove excess particles in the most preferred angles, but it didn’t help.