Hi CS users,
I have seen many great advices from this forum but I could not find an exact solution to my problem, I am asking for helps…! This is a long story, but I thought it will be better to share details.
I am allowed to upload just one media, so I attach one very long picture with labels. (You may have to browse the picture in a separate window, sorry for the inconvenience)

I am trying to get a structural model of a relatively small protein, which is about 90 kDa.
The protein I am actually dealing with is even smaller (<50 kDa) so a Fab is used to make it bigger, to ~90 kDa.
The data were collected using Krios 300 kV with Gatan K3 detector.
Pixel value is 0.664 A/pix and 24,000 movies were collected.
From this data the processing procedure was as follows:
(Parameters were set to default, otherwise stated)
- Full frame motion corrected and patch CTF carried out
- Blob picker
- 2D classification to get templates
- Template based particle picking was done and initial particle number was 11.7 M particles
- Several rounds of 2D classification were carried out and left with 3.4 M particles (showing only classes of different views) F1
I am aware of the orientation bias, where the side view is not exactly the side but just a bit of angle around from the front view. To solve this problem, I prepared grid in different way and found right condition where I could get this F2. However this is from screening and I am planning to get data for high resolution processing later.
However, another problem comes later with this current data.
I will continue with my procedure:
- From 3.4 M particles I made a subset with 1 M particles and I ran ab-initio reconstruction and I get these models F3.
I can see clearly that class 1 seems as my model (891,000 particles) F4.
So I tried next steps:
- Heterogenous/Homogenous/Non-uniform Refinements
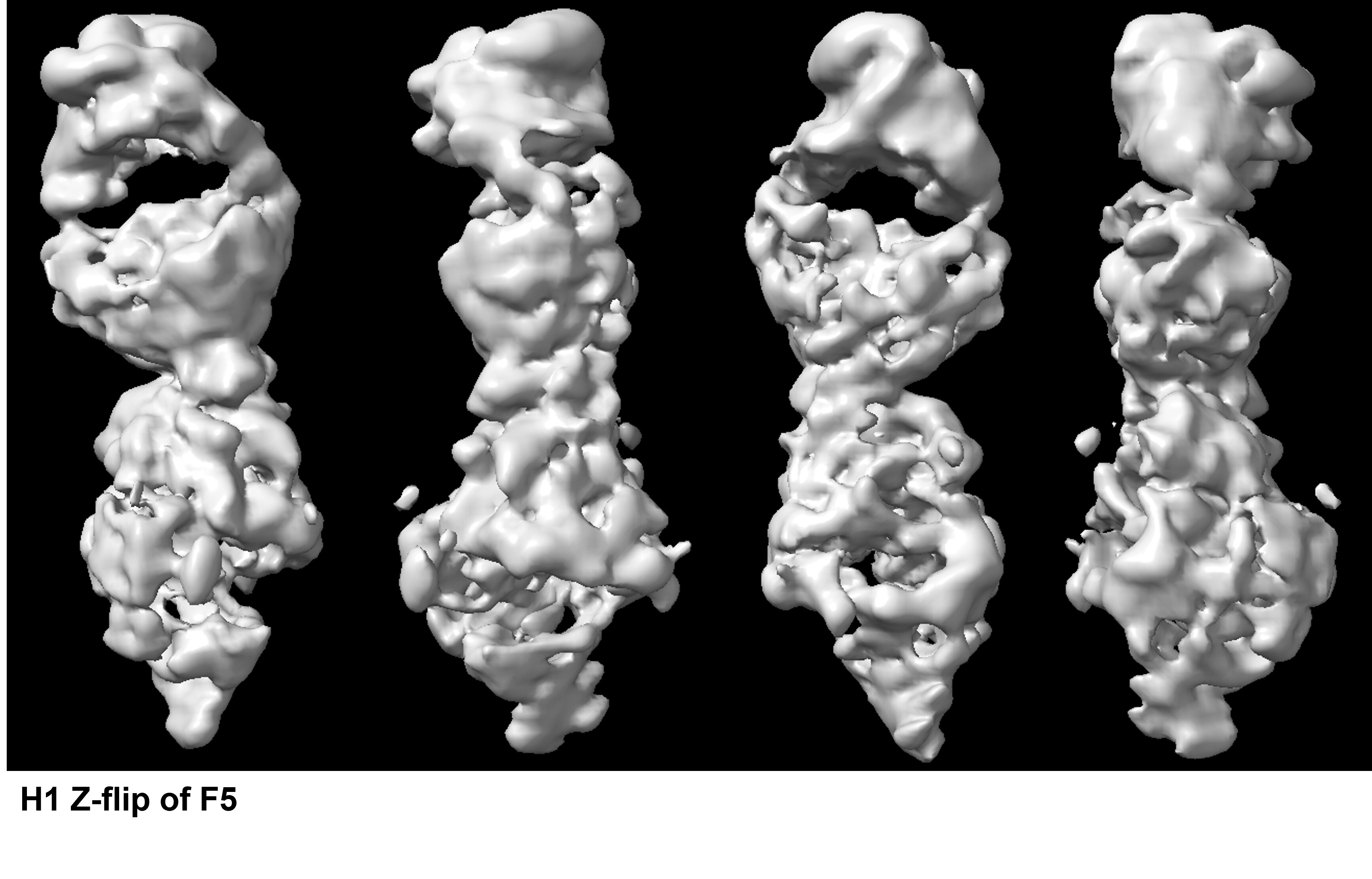
The NU-Refinement showed the best results but I could see streaks (which may have come from the missing views) and the model is no where near as a real structural model. F5 and F6.
Apart from the problem probably caused from the missing view, when I checked what particles were used to reconstruct the model above I got this F7.
Out of 891 K particles, only about 10 K particles are the top view. Also, these top view like particles look so bad compared to the top views I saw with 2D.
Then I could deduce three conclusions:
- The top view I thought is not actually the top view - which I think is very unlikely because the secondary features I saw from 2D are true.
- Top view could not align because there is missing side view - this also seems not so likely because with the screen data where I got the new side view, it still shows ab-inito reconstruction in similar outcome and particles were also checked after 3D reconstruction and showed a very similar results - very little number of top views.
- Maybe, and hopefully it is area of processing where there is some parameters I could change with to actually let top view to fit into rest of the views.
(Plus, I ran many more rounds of 2D classification to really clean up the particles, where I ended up with about 850 K particles - which showed the same results as above)
(Also, I tried manually fixing the ratio of views to 1:1 but it gave a very weird model and when I do ab-initio reconstruction with this ratio adjusted particles and one class only, the half of the particles go to “unused particle” which is probably the top view)
I will appreciate a lot if there are any tips or answers to this problem…!
Thank you
Seong