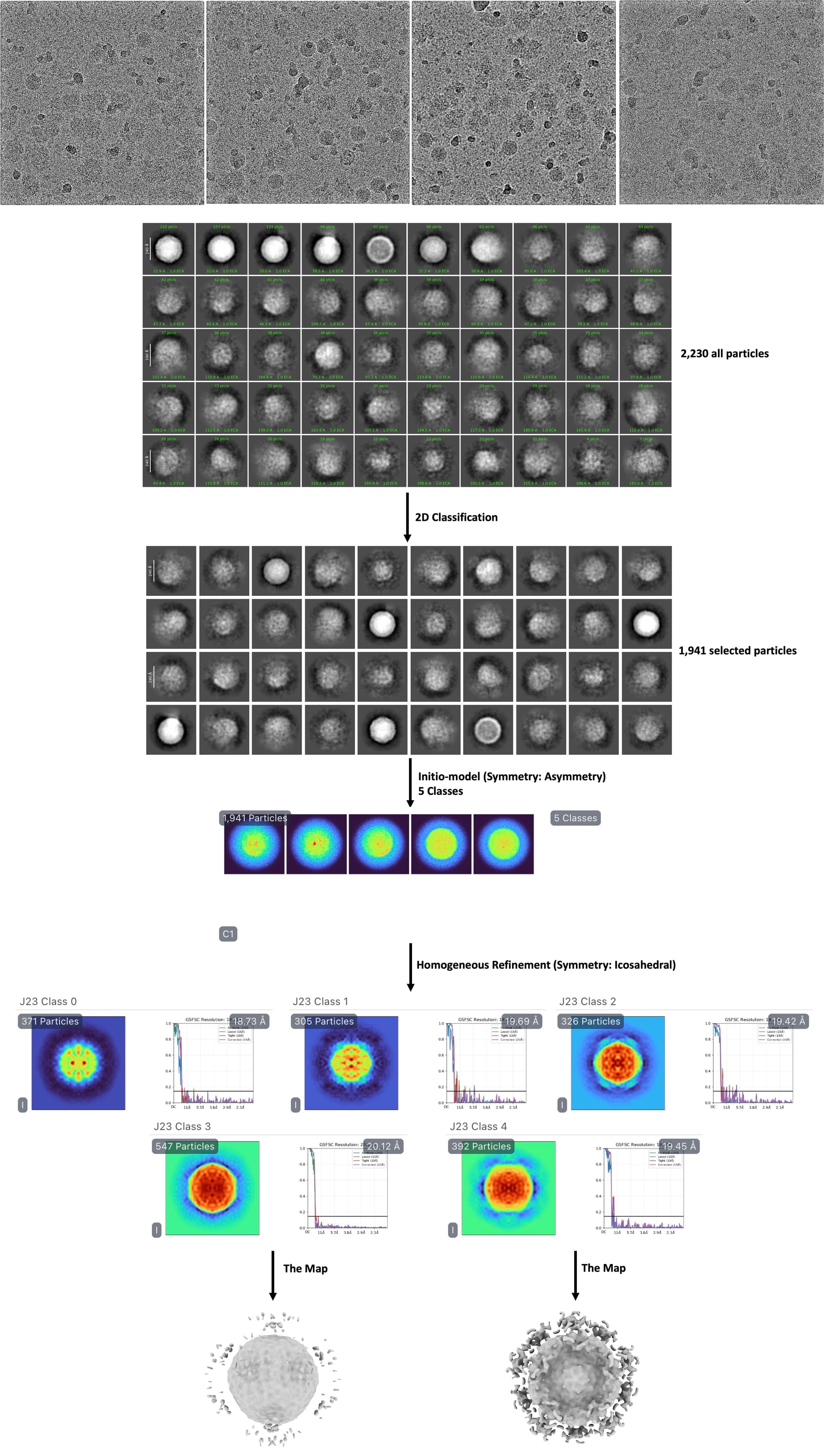
I collected over 2,000 micrographs at 200 kV as a preliminary test to optimize my techniques before proceeding with large-scale data collection at 300 kV. During data processing, I used cryoSPARC’s template picking to select 5,277 particles from a mixture of EVA71 virus and SCARB2 protein, with the goal of reconstructing the binding complex map.
However, analysis revealed that approximately 80% of the EVA71 particles were not bound to SCARB2, while only about 20% showed binding - though I suspect many of these bound particles are not fully saturated with SCARB2.
My current processing workflow consists of:
Performing ab-initio reconstruction with C1 symmetry, classifying all particles into 5 classes
Conducting homogeneous refinement with icosahedral (I) symmetry
The main issue I encountered was that during averaging, the extra density on the viral surface (representing SCARB2) became averaged out, resulting in a mask that only captured the unbound virus particles. Consequently, the receptor density disappeared from the final reconstruction.
According to references, successful reconstruction of the EVA71-SCARB2 complex map (EMD_0332) requires about 10,000 particles. The EVA71 capsid exhibits icosahedral symmetry, while SCARB2 binds as a dimer.
Another critical point to note is that EVA71 particles become highly unstable upon SCARB2 binding, undergoing rapid degradation. This explains why many virus particles in our dataset appear aggregated or ruptured.
Given these challenges, I would greatly appreciate expert advice on how to optimize my approach to obtain a high-quality map of SCARB2-saturated EVA71 complex. Attached is a schematic of my current analysis pipeline for reference.
Thoughts from a very quick search regarding both EVA71 and SCARB2…
You’re unlikely to get every possible binding site on the capsid with the receptor bound, simply from steric hindrance, SCARB2 is not small.
– Therefore symmetry imposition may not be the best idea.
– Conformational change should also be considered, and indeed degradation of the virus as you mention.
– You’d need something from 1:50-1:10,000 virus:receptor to stand a chance of every site demonstrating binding, but the sheer size of SCARB2 will make that impossible.
In this situation I’d try the following: pick every class which isn’t junk, single class I1 refinement, symmetry expand, use a very, very soft mask on one region/binding site and 3D classification into a fairly large number of classes at moderate resolution (maybe 8-12 Å). Then local refinement of best class (due to symmetry expansion; also global refinement will almost certainly lose the SCARB2 alignment against the capsid).
Thanks for your reply.
Regarding Those low-resolution particles that look like junk are mostly lysed viruses, because once the receptor binds to the virus, it will lyse the virus very quickly. I just tried your method and found that after excluding these broken viruses, the extra density becomes less. Through 3D classification, I can’t see obvious differences between classes, so I merged all expanded particles together and used a very very soft mask to perform local refinement on the binding site, but the results weren’t ideal.
As for whether SCARB2 saturating EVA71 would cause steric hindrance, I think it should be fine - i think as long as we have enough bound particles, we should be able to reconstruct a SCARB2-saturated complex like EMD_0332.
But I have a question: since the virus itself is icosahedral while SCARB2 usually binds EVA71 as a dimer, how should we best handle this symmetry mismatch? I believe using I1 symmetry in homogeneous refinement will alter the true symmetry of the SCARB2 density.
Actually, I don’t think the strategy of trying to maximize SCARB2 binding to EVA71 is optimal, because the interaction between SCARB2 and EVA71 appears to be quite flexible. When picking particles, it’s very difficult to visually identify SCARB2 density in the micrographs—it only becomes apparent after refinement and 3D classification. However, I believe many EVA71 particles likely have SCARB2 bound, just rare or not saturated.
Is there an alternative approach, such as:
First, performing local refinement to obtain a moderate-resolution map of the SCARB2-EVA71 binding site (even if the resolution isn’t high).
Then, using this map as a template to search for similar binding sites across all micrographs?