Hello all,
I am processing a dataset for an icosahedrally symmetrical particle with a small peptide bound at *theoretically* every asymmetric unit, and am having a difficult time resolving it to the same degree as the icosahedron. My refinement steps so far (heterogenous, homogenous, non-uniform) have icosahedral symmetry imposed, and I have a conceptual question regarding imposing symmetry when there is a non-symmetrical feature in the particle (the peptide in this case). If the peptide has full occupancy across all binding sites in each rotational symmetry group, would it be considered an “extension” of the symmetry of the whole particle? Or is there a possibility that it would get averaged out since it is not a symmetrical feature of the particle, per se? Hope my reasoning makes sense, thanks in advance!
Flow
If the binding is the same at all sites, it could be, yes.
There is a distinct possibility that not all possible binding sites are occupied. Or the binding site is flexible enough to cause the same effect. Or the peptide is flexible enough for the same. Unless you are adding probably ten- or even hundred-fold more peptide than particle (relative to all potential binding sites being occupied, so potentially already 60x more peptide than particle), partial occupancy is quite likely. And peptide flexibility is almost certain.
True rigidity in proteinaceous constructs is vanishingly rare (impossible, if approaching atomic resolutions (b-factor).  Even the most rigid icosahedral particle will be subtly flexing around. Sometimes this is obvious (difficulty in resolving flexible P-domains in capsids when compared to the more rigid S-domain) and sometimes it is more subtle. Allow me to hypothesise for a moment… if the flexibility were in the Z-axis (i.e. parallel to the beam) it should only be biological flexbility, even if mechanical in origin (e.g., maybe very thin ice compresses the particle slightly, or solvent/air interface contact does the opposite)… but if in the X- or Y-axes, until extremely high resolutions it might be difficult to identify if it was biological particle distortion (i.e. flexibility) or electro-optical aberration (i.e. magnification anisotropy, although this should be uniform across a given beam tilt, and thus alignment and back-calculation should identify contributing factors here).
Even the most rigid icosahedral particle will be subtly flexing around. Sometimes this is obvious (difficulty in resolving flexible P-domains in capsids when compared to the more rigid S-domain) and sometimes it is more subtle. Allow me to hypothesise for a moment… if the flexibility were in the Z-axis (i.e. parallel to the beam) it should only be biological flexbility, even if mechanical in origin (e.g., maybe very thin ice compresses the particle slightly, or solvent/air interface contact does the opposite)… but if in the X- or Y-axes, until extremely high resolutions it might be difficult to identify if it was biological particle distortion (i.e. flexibility) or electro-optical aberration (i.e. magnification anisotropy, although this should be uniform across a given beam tilt, and thus alignment and back-calculation should identify contributing factors here).
Hi Flow,
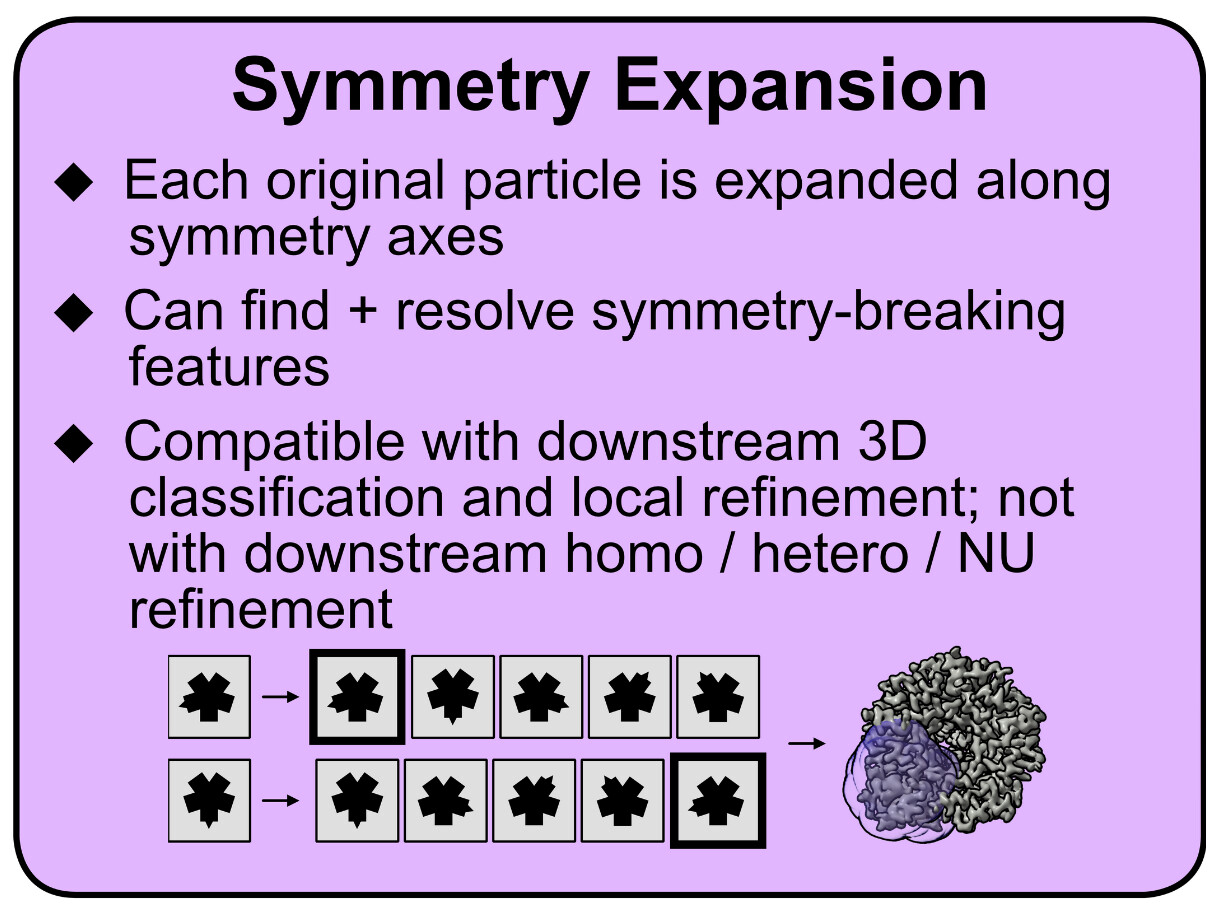
In addition to @rbs_sci’s excellent reply, if you are trying to actually resolve this feature, depending on the size and why exactly it is not resolving well (i.e. occupancy vs. flexibility vs. slightly different binding modes), you may be able to use symmetry expansion for this (I copied part of a teaching material I made on this below). Basically, after expansion, you can use 3D variability and/or classification to group symmetry-expanded particles together that either have yes/no occupancy in a certain area, or have the peptide bound in the same way. Figure S2 in this paper (Shah et al 2024, Structural basis of VCP-VCPIP1-p47 ternary complex in Golgi maintenance) also has a nice visual description of this in practice.
Thank you both @rbs_sci and @tlevitz for your insights! I will consider doing symmetry expansion with followup local refinement to see if that improves things.