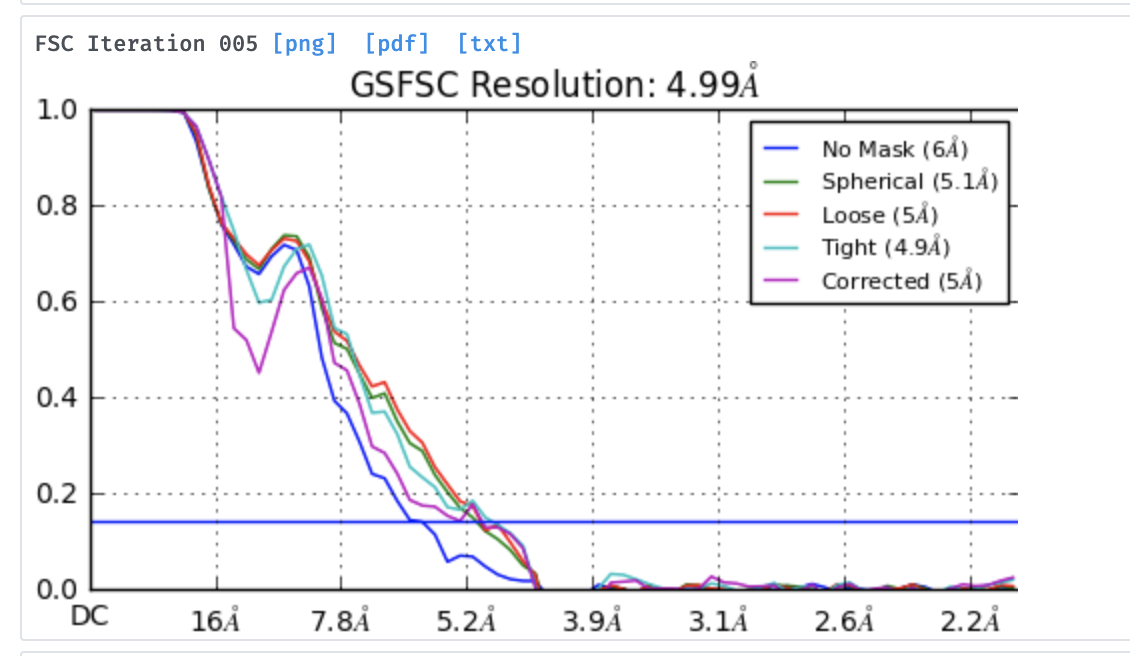
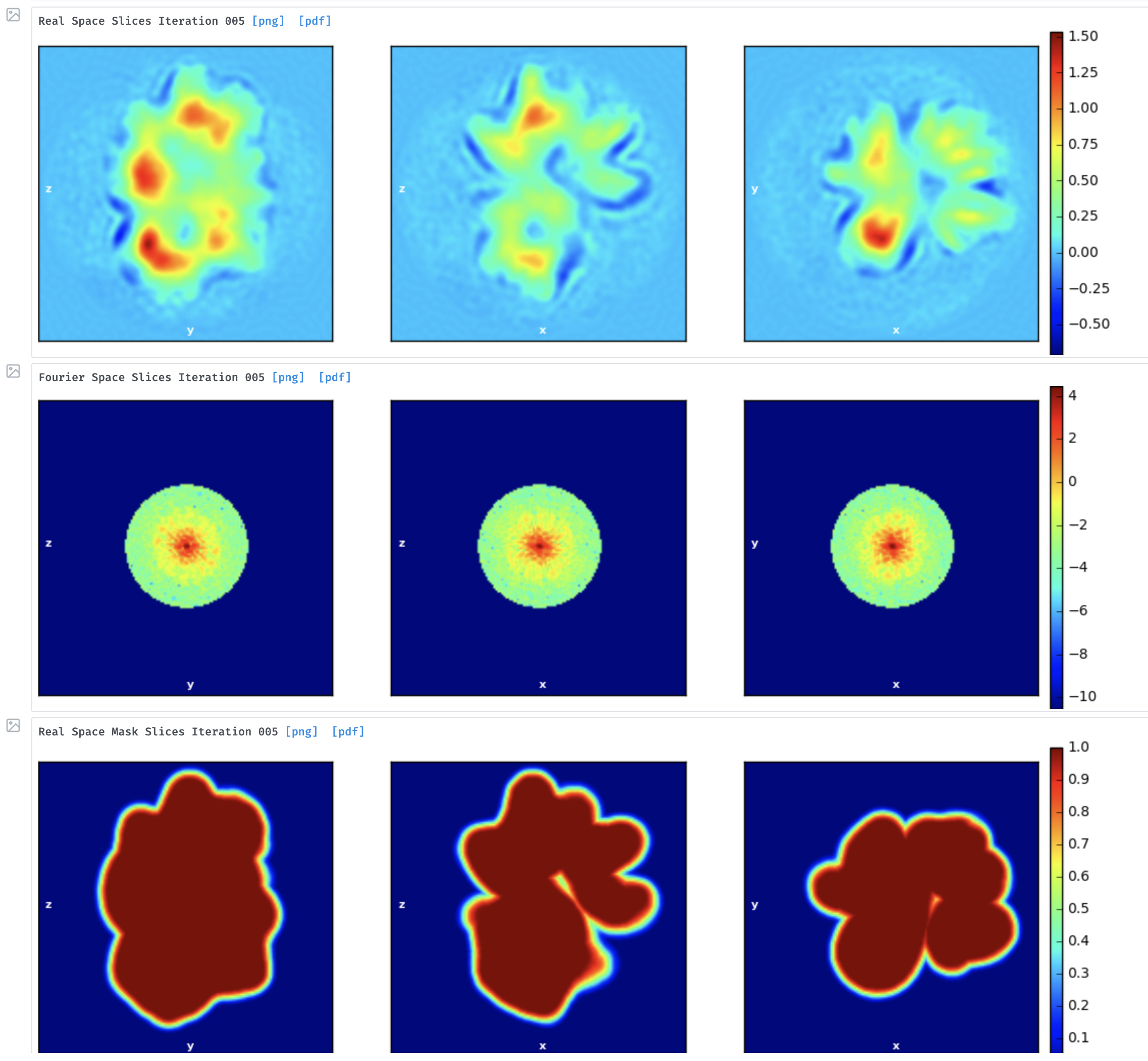
After doing homogeneous refinement of the selected models in cryosparc, I’m seeing a major discrepancy in the GSFC resolution reported and the resolution of the map that I download after refinement. For example, in once case I had the reported GSFC resolution to be ~5 angstroms but the map is definitely not at par with the reported resolution (see images attached). I’m wondering if I’m doing anything wrong during refinement, which is resulting in such a huge gap between the reported vs the actual resolution. Thanks!
Hi @ketanm thanks for reporting. This is definitely a strange result (and not near 5A)
There are a few questions that come up at this point:
How many particles?
What did the 2D classes look like?
How did you do particle picking? Is there any chance you may have “double-picked” the particles or imported the micrographs twice? If the same particles are appearing on both sides of the gold-standard split for some reason, that would cause behavior like this
Did you create that mask by hand?
The box appears to be cut very tight around the protein. Generally speaking, the box should be about 1.5x -2x larger than the particle
Where did the CTF estimates for this data come from? What were the CTF fit scores?
Were any parameters in refinement changed? Especially the initial lowpass resolution?
I would say that you should first try to re-extract the particles with a larger box and re-do the refinement.
How did you do particle picking? Is there any chance you may have “double-picked” the particles or imported the micrographs twice? If the same particles are appearing on both sides of the gold-standard split for some reason, that would cause behavior like this Quite certain that particles weren’t picked twice. Used cryolo with a general model and not trained by us. We extracted particles using Relion 3.1 and then imported them into cryosparc.
Did you create that mask by hand? No
The box appears to be cut very tight around the protein. Generally speaking, the box should be about 1.5x -2x larger than the particle Particle size is 150A & we used a box size of 156A since the particles are very crowded but we will definitely try a bigger box size and get back to you
Where did the CTF estimates for this data come from? What were the CTF fit scores? Ctffind4 ran as a wrapper in Relion3.1. CTF fit score aka Ctf FOM ranged from 0.01-0.2
Were any parameters in refinement changed? Especially the initial lowpass resolution? Used default initial low pass of 30A
Hi,
I would like to add that I also see this overestimation of resolution quite frequently in data sets that are more difficult (small or heterogeneous particles), when all processing is done internally in cryoSPARC and using the default settings. Using real space masks with smoother edges compared to the default or increasing the GSFSC split resolution somewhat helps, but not entirely. I will be happy to hear if there are other suggestions.
Additionally, the real space slices presented here by @ketanm show a “dip” in density around the map followed by stronger density at the periphery. This creates sort of an artificial shell around the map, which I often see in such cases as well. I wonder if this relates to the overestimation of resolution and how can you avoid it (apart from having a much smoother mask)?