Hi CryoSPARC Community,
I am currently working on a cryogenic EM dataset of a 12-transmembrane (12-TM) helix protein in DDM detergent. The molecular weight suggests a potential homodimer, and the protein contains an extracellular domain (ECD) predicted to be highly flexible.
I have hit a major bottleneck during downstream refinement and would appreciate your insights on whether this dataset is salvageable and how to optimize my workflow.
What I have done so far:
-
2D Classification (Fig. 1): The 2D classes show clear transmembrane helical tracks/striations and characteristic micelle densities, though the overall orientations appear somewhat limited and the flexible regions add blurriness.
-
Ab-initio Reconstruction (Fig. 2 & 3): I ran Ab-initio with 6 classes using a relatively aggressive resolution cutoff to capture helical features:
-
Maximum resolution: 7 Å | Initial resolution: 12 Å | Fourier radius step: 0.01
-
Initial / Final minibatch size: 300 / 1200
-
Result: Among the 6 classes, Class 003 (shown in Fig. 3) successfully resolved a promising TM region that looks structurally plausible and well-defined.
-
-
Heterogeneous Refinement (Fig. 4): When I input this promising Ab-initio class into Heterogeneous Refinement or Non-Uniform (NU) Refine, the resolution stalls. Instead of resolving the protein, the density of the DDM micelle becomes progressively sharper and stronger, while the TM helices become blurred or features get washed out. It appears the alignment is heavily driven by the spherical/ellipsoidal shape of the DDM micelle rather than the protein itself.
My Questions to the Community:
-
Feasibility Assessment: Given the clear helical features in 2D and a plausible TM zone in Ab-initio, is this dataset worth pursuing further, or is the alignment over-fitting to the micelle too severe to overcome?
-
Ab-initio Validation: Does the obtained Ab-initio volume look trustworthy, or could the apparent “TM feature” be an artifact driven by the eccentric shape of the DDM micelle?
-
Mitigating Micelle Dominance: What specific strategies can I use to prevent the strong, low-resolution DDM micelle signal from dominating particle alignment in NU-Refine? (e.g., custom masking, modifying the initial low-pass filter, or adjusting per-particle scale optimization?)
Any advice on masking strategies, parameter tweaks, or alternative workflows would be immensely helpful!
Thanks in advance for your time and help.
Best regards, JF Huang

Figure 1
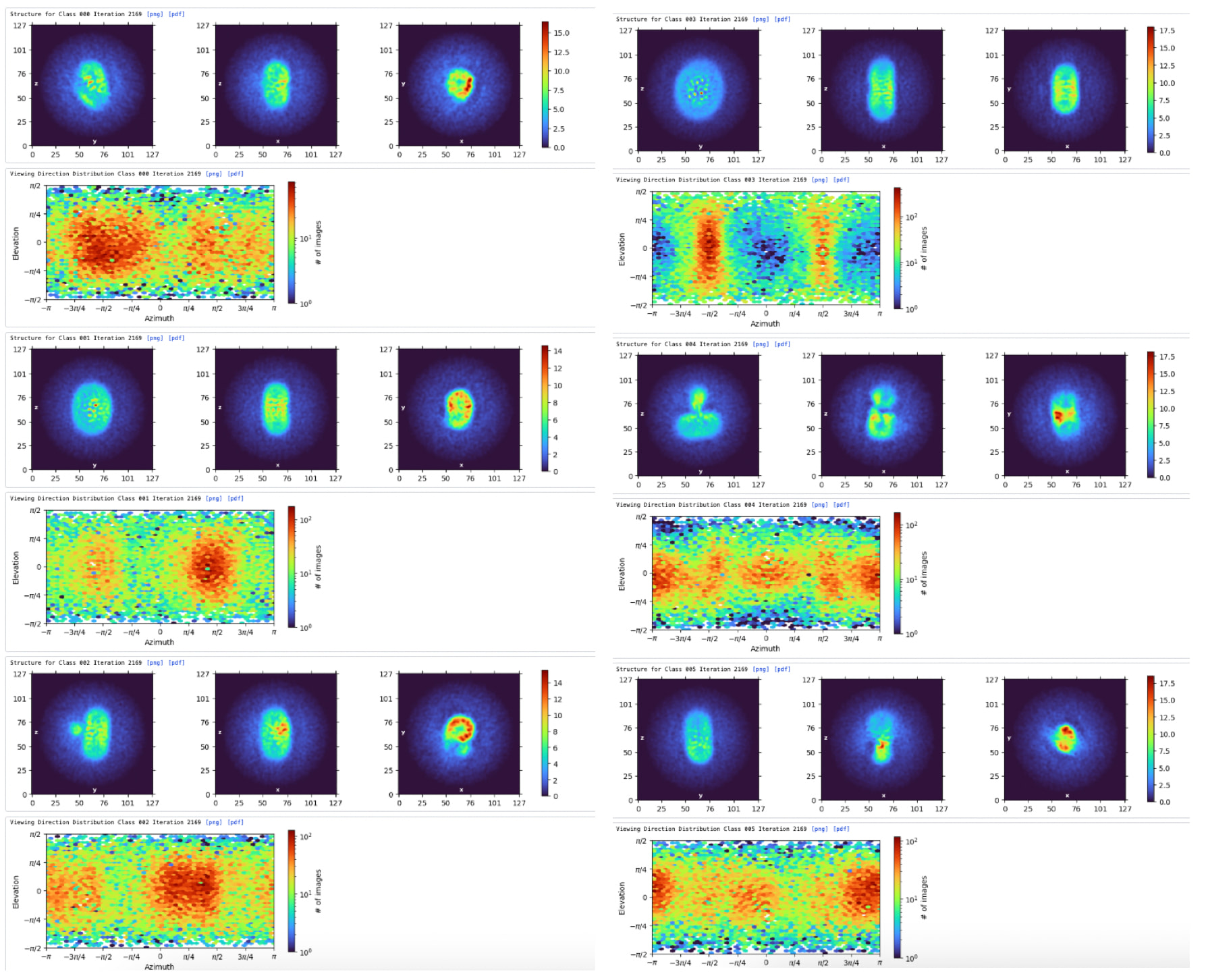
Figure 2
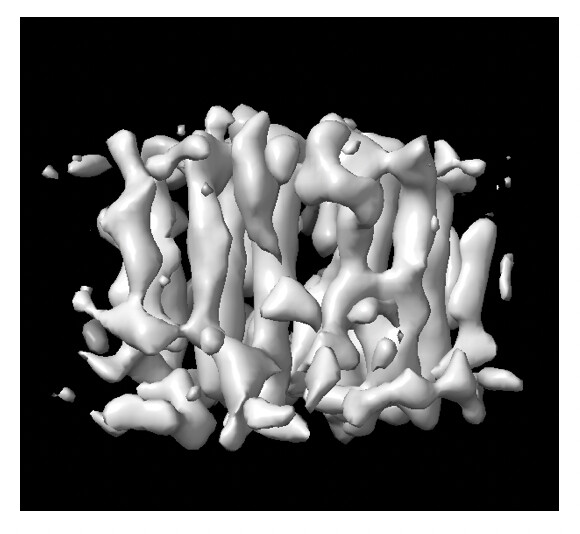
Figure 3
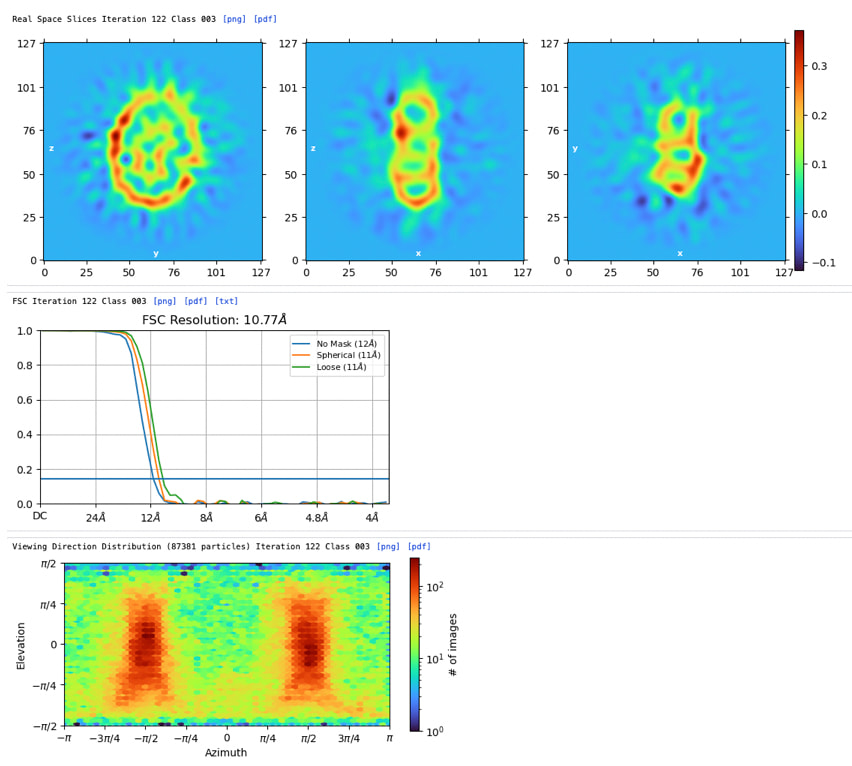
Figure 4