Hi everyone,
I’m relatively new to cryo-EM image processing and would appreciate some advice on a challenging membrane protein dataset.
The target is a native membrane protein complex, purified in 1% DDM + 0.1%CHS, then exchanged to 0.02% GDN. The sample appears highly heterogeneous, and the exact composition/oligomeric state is unknown. Based on biochemical data and AlphaFold3 predictions, I expect the particle to be around 15 nm, although it could potentially be somewhat larger.
I have also processed the dataset in RELION, but unfortunately without better results.
Dataset
-
~23,000 micrographs
-
Pixel size: 0.74 Å/px
-
Initial blob picking:
-
Minimum diameter: 90 Å
-
Maximum diameter: 200 Å
-
-
~6 million particles extracted
-
Box size: 384 px
-
Fourier cropped (binned) to 192 px
Initial processing
To make processing manageable, I split the dataset into batches of ~500,000 particles and performed multiple rounds of 2D classification on each subset (parameters attached in screenshot).
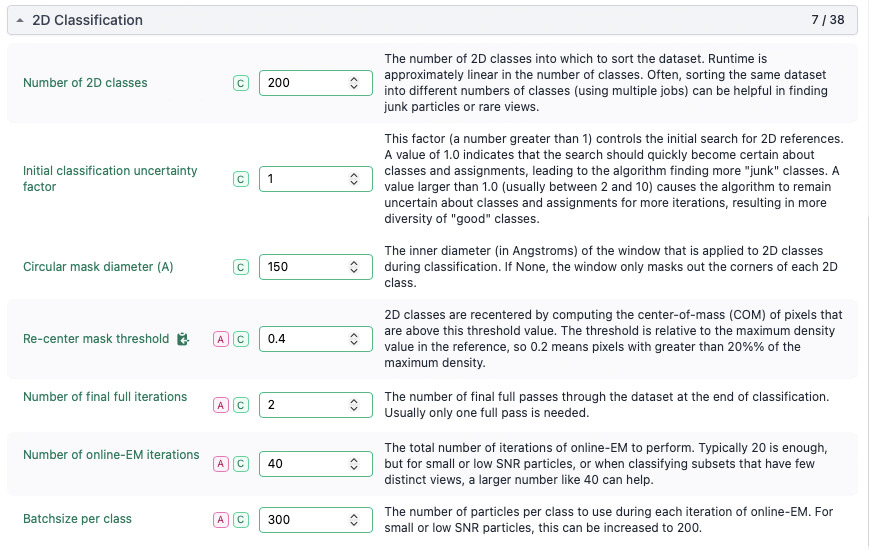
After selecting reasonable classes, I merged the selected particles and performed additional rounds of 2D classification.
The resulting workflow was roughly:
Blob picker → Extract → Multiple rounds of 2D classification on subsets → Merge selected particles → Additional 2D classification
Template generation
I then generated ab initio volumes. Heterogeneous refinement and Non-Uniform refinement did not produce any high-quality maps, so I used one of the ab initio volumes that appeared most promising as a template (screenshot attached).
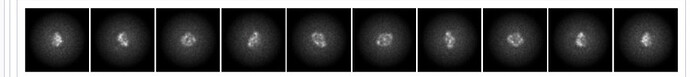
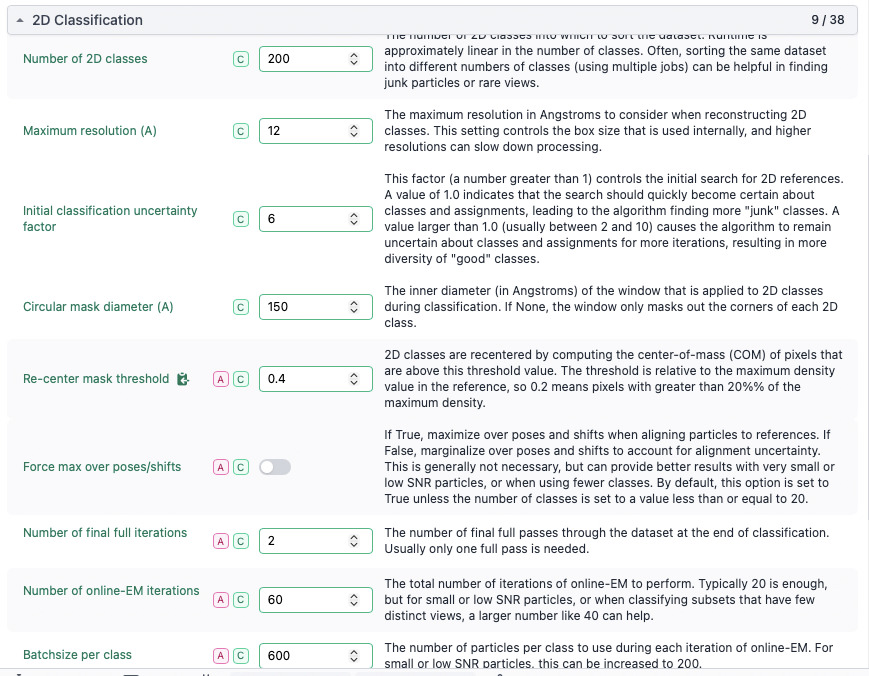
I performed template picking using these volumes and repeated 2D-classification workflow with changes parameters (screenshot attached).
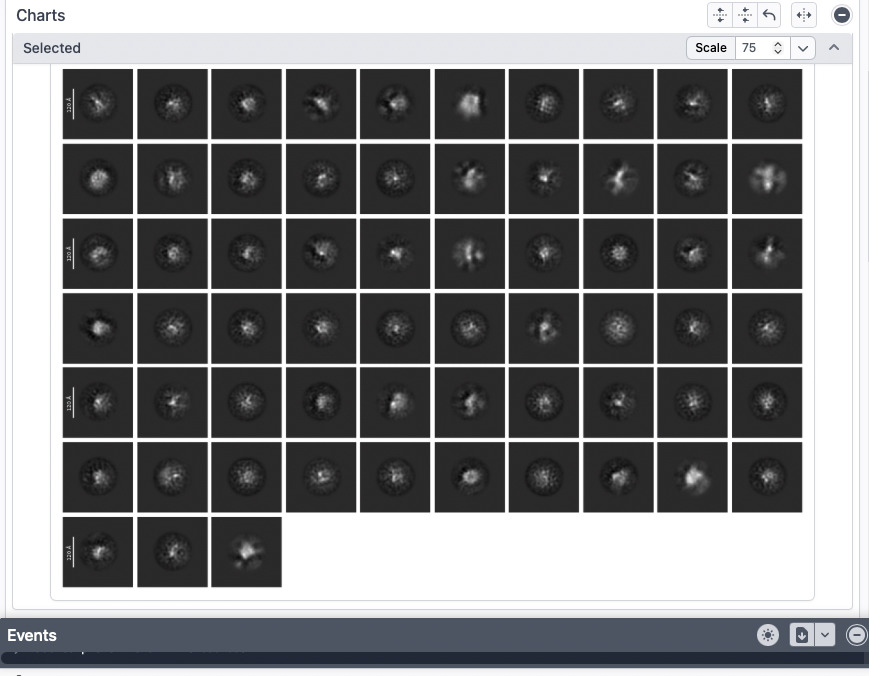
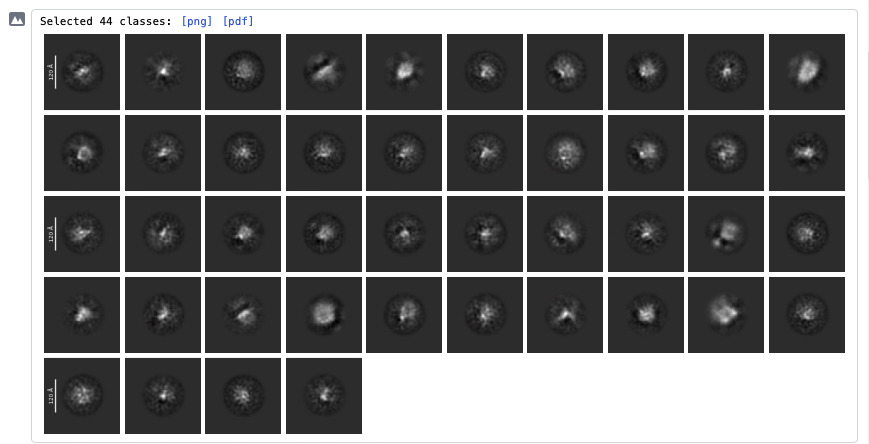
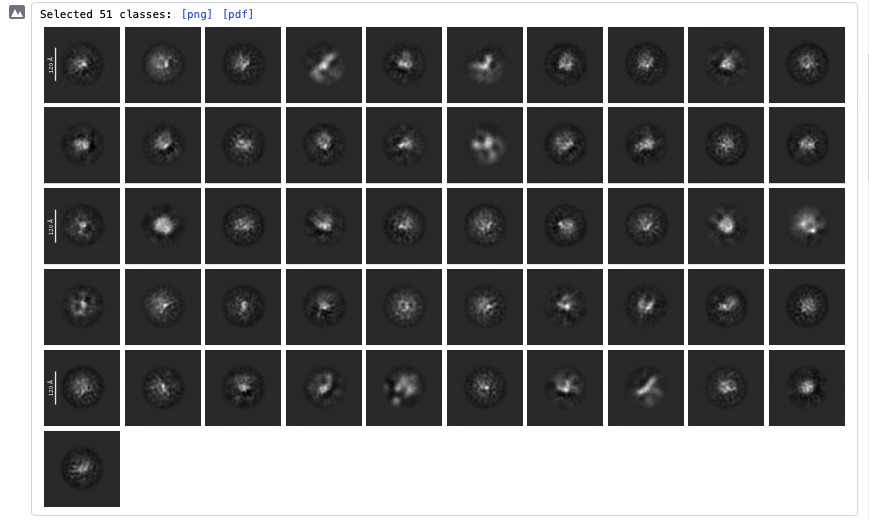
Current results
Some of the 2D classes look potentially interesting (attached screenshots). A few classes appear vaguely consistent with features expected from my AlphaFold3 model, but honestly it is difficult to say with confidence.
I also tried using AlphaFold3 model for template picking, but this did not improve the results.

Questions
-
Does this overall workflow seem reasonable for a highly heterogeneous membrane protein sample?
-
Are there any obvious processing steps or parameters that you would change?
-
Would you recommend trying alternative approaches/software (Topaz, CryoDRGN, 3DVA, ISAC, etc.) at this stage?
-
Is there anything in the attached 2D classes or ab initio volumes that suggests I may actually be looking at real particles rather than noise/contaminants?
-
For a difficult membrane protein dataset like this, what would you try next?
Any feedback or suggestions would be greatly appreciated. I’m still learning and would be very grateful for an assessment of whether I’m following a sensible workflow or heading in the wrong direction.
Thank you!
