Hi all,
could you please give me an advice how to improve my 2D classification? It looks like 2D classes are not improving. Dimeric complex is quite nice, I am able to see even secondary structures in 2D classes but trimeric is a huge problem (see picture). Dimeric is structured and stable, but third protein is quite flexible. Do you have please please some advices what to do with that? I am starting to be really desperate  . I know, the amount of particles is not so high so we will collect bigger dataset but this happens all the time, even when we had dataset about 130 000 particles. Thank you so much for all ideas!!! Have a nice day
. I know, the amount of particles is not so high so we will collect bigger dataset but this happens all the time, even when we had dataset about 130 000 particles. Thank you so much for all ideas!!! Have a nice day
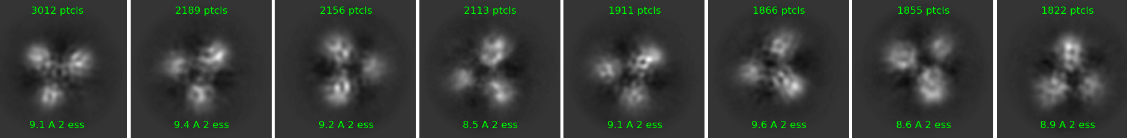
How are the classes balanced? If you have very little trimeric particles, you could try to pick specifically on the trimeric particles, and make 2D classes with only trimeric particles. Or alternatively rebalance your classes (there is a job for it in Csparc) and then try from there.
Another possible problem is that the trimeric particles are just slightly oversized for your boxsize/circular mask. Adjusting that might help.
Good luck!
Jitske
THANK you so much! I will try all of your suggestions…especially I did not know that there is class about rebalancing of classes.
Have a nice day!
Alžbeta
if dimeric assembly is actually very stable, then it should look good in 2D with a fuzzy 3rd part. might not be the case. 2D is also wonderful to see particles and sometimes sort them, but it is not required. Can skip to 3D if ab initio looks good. It will be somewhat difficult for the program at all steps since you “particle” of 3 domains has no strong center of mass.
Yes, 2D is nice with visible secondary structure and when I try to pick it is simple and 2D are beautiful. So I do don understand why we are not able to see stable dimer with addition protein. Do you have any suggestions how to improve 2D classes? I would like to skip to 3D but tutorial it says that I have to put imput there particles from refinement homog but when I am not able to classify 2D then I am in a never ending circle… or do I understand it in wrong way?
Thank you!
if two different samples/datasets behave differently, even if unexpected, it is probably telling you something. third protein breaks apart the first two, or dataset is less high quality.
you can skip 2d. for ab initio you need particles only - can be from import. for 3D NU-refine or homogeneous refine you need a 3D volume reference to go with the particles - but this could be from ab initio. none of this needs 2d. 2d is usually best place to make sure picking went well, remove any of the OBVIOUSLY wrong picks, and keep almost all particles which look decent. that being said: if you can’t get good looking 2D classes then you will probably have a very hard time getting nice structures.