Hello Everyone,
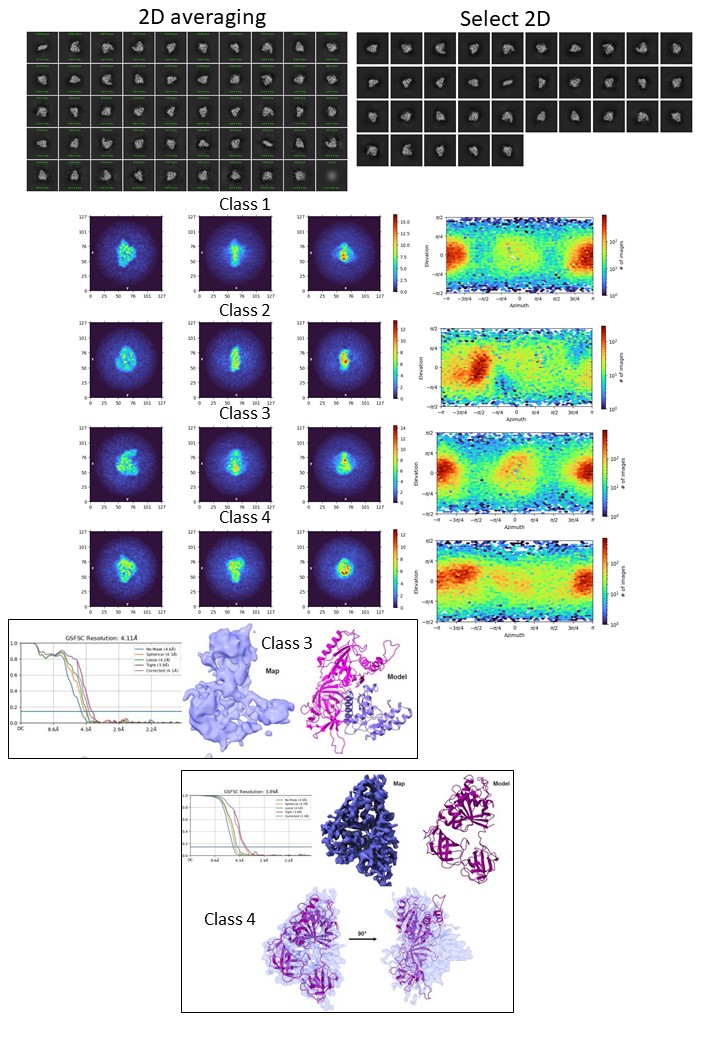
I am struggling with a protein of Mw 120 kDa. The data was collected using Krios 300 kV at 105kX mag and 0.85 A pixel size. The particles are extracted in a box size of 256 pix. After multiple rounds of 2D, the particle seems to be aligned. The resulting particles are 3.3 million which were subjected to 4 ab initio classes (parameters changed: initial res 12, final resolution 8, C1). From the ab initio, class 2 was omitted from the homogeneous refinement as it is a domain we identified from the complex that broke off. The rest three classes were refined with default parameters of homogeneous refinement as non-uniform refinement worsened. Now the problem is that although the model fits into the volume, the secondary features hardly align. I have tried heterogeneous refinement to further clean up the particles but the final map stays uninterpretable. I would be happy if somebody could help me out with this.
Thank you.
If you still have 3.3M particles, you have a lot more room to go nuts on cleaning in both 2D and 3D. First thing I’d suggest is massively increase the number of classes you use in 2D - 200-400 perhaps - and play with parameters like uncertainty factor (I like 5), non-negativity, etc. Also, try to set enough particles per class, and enough iterations that you pass over the full dataset at least once (preferably twice) during the subsets, and maybe use 3 full passes to finish.
The 2D looks promising, but my suspicion is that there is a lot of junk still hiding in there, which is killing the 3D.
edit: Also, looks like a strong orientation preference. Cleaning it up and dumping some extraneous classes might help, or play with detergent additions or stage tilt with a future acquisition (if necessary).
My suggestion would be to clean the data set much more, you still have >3M particles. Coudl you share your 2D classification parameters?
Also, make sure that got the map’s handedness correctly.
Sorry, its not 3.3 million instead its 0.33 million.
Hi Dario,
Thanks for the suggestion. In the previous 2d classifications i used 100 classes with 400-800 batch size. Iterations from 50-75. Force max off. Rest all default.
I did the volume flip(z) in chimeraX and fitted the model from ab initio classes, but it does not improve.
@rbs_sci although the azimuthal shows preferred orientation, i see lot of views. Is it possible that the software is unable to reconstruct as the x and y and z orientation of the protein are quiet different?
how many in Class4? het refine particles from 1,3,4 using 5 volumes of class4. Though you’re sub-4Å, I agree there are contaminating junk particles. another way to clean up is to keep only those particles from micrographs of relatively good CTF estimation now that you already have them assigned to one structure (using the low-resolution information that is constant for all particles). Basically cleanse the class 4 of it’s lowest resolution particles, which is hard to do with other jobtypes.
About that, did you do exposure curation? Try to keep only the absolute very best ones in terms of ctf fit, relative ice thickness and Astigmatism, and extract the assigned particles from the curated micrograph Set and see if that improves anything.
Your 2D classification parameters seem fine. You could still play with mask, but the averages look decent already, supporting the idea that the data set is still contaminated with junk. Try running several more 2D select/2D classification iterations.
I think I agree with everything that has been said, just not sure about how much junk you do have, or the problem is simply the flexibility of the protein. Before cleaning further, you can try 3DFlex on the full clean set, and also on classes 3 and 4. Other jobs to try are 3DVA and 3D classification with different parameters; but I’d start with 3Dflex. With so many particles you are in the lucky side.
@DarioSB Yes i did an exposure curation and kept the micrographs with CTF better than 5A, Ice thickness 1.1 cut off and did not change the Astigmatism.