Dear all,
I am writing about a problem we encountered during the reconstruction of a 150 kDa protein complex using CryoSPARC.
In total, around 19,000 movies were collected, and approximately 1.5 billion particles were picked using Topaz Train, resulting in the 2D classes shown below. The 2D classes appear to show secondary structure details, but also indicate a preferential orientation of the particles.
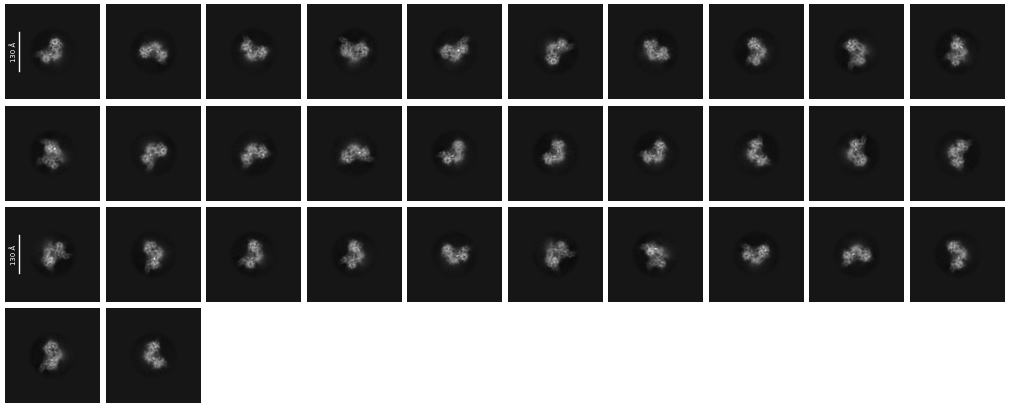
The core of our complex is about 120 Å in diameter and consists of a tetramer of one protein, along with up to four molecules of another protein. After 2D classification and ab-initio reconstruction, we performed particle cleanup, resulting in ~500,000 particles after the first particle sorting.
Using these particles, we performed an initial non-uniform (NU) refinement, which produced an FSC curve showing a bump around 4–5 Å.
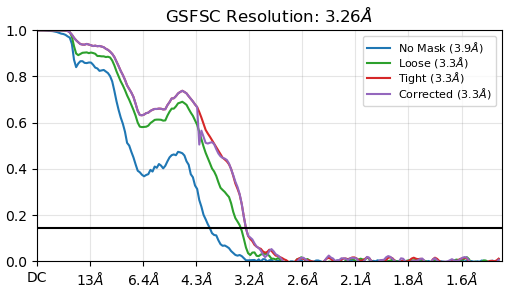
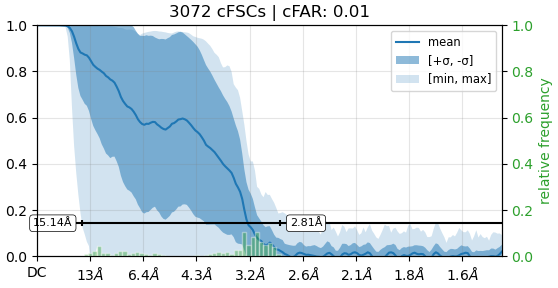
Upon closer inspection, only the central part of the map shows reasonable density, while the left and right parts appear somewhat random, featureless, and fragmented — preventing reliable model building.
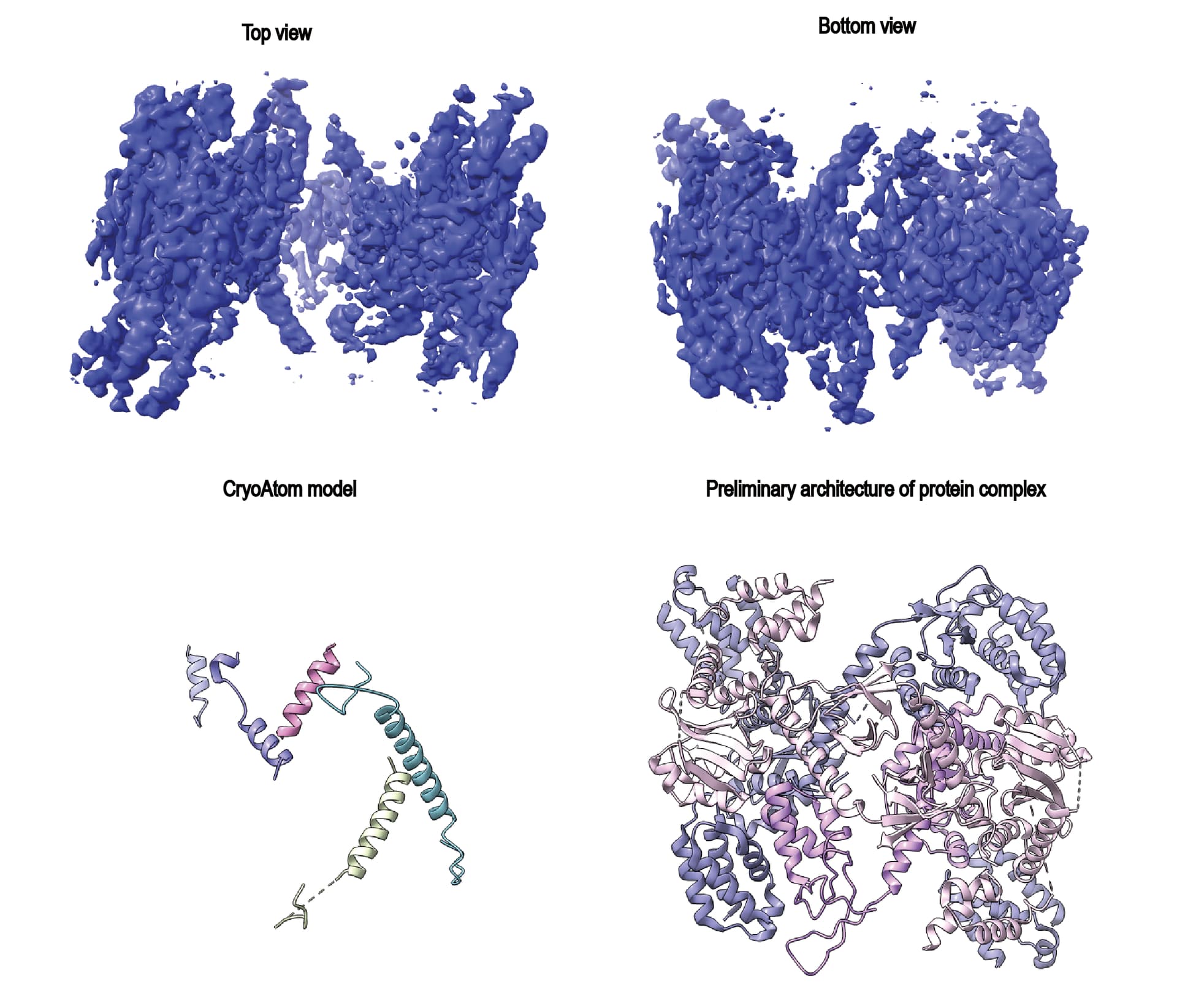
We then attempted further particle sorting using heterogeneous refinement, orientation diagnostics, and orientation rebalancing, but obtained essentially the same result. The density did not improve even when changing the box size (from 300 px to 440 px, pixel size 0.73 Å).
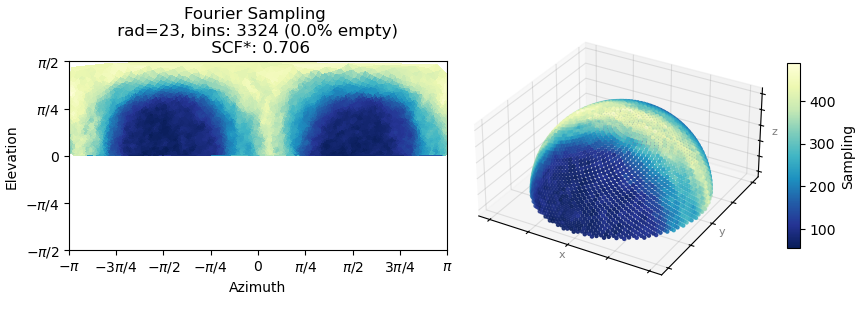
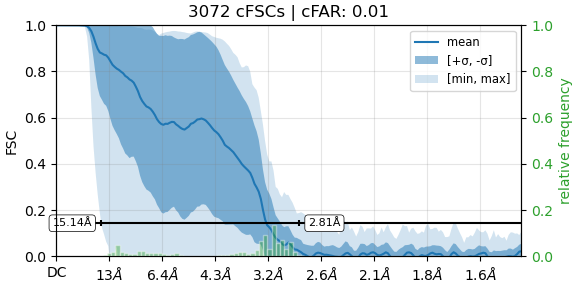
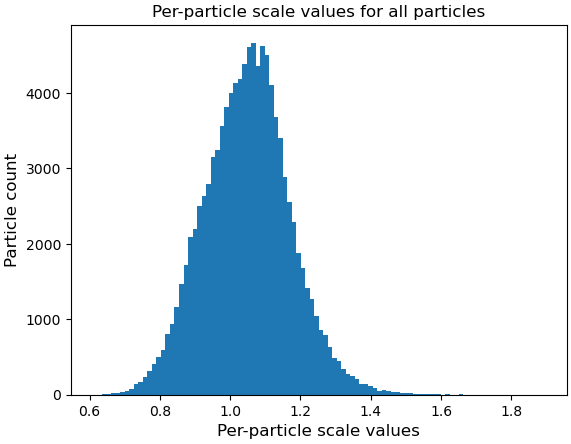
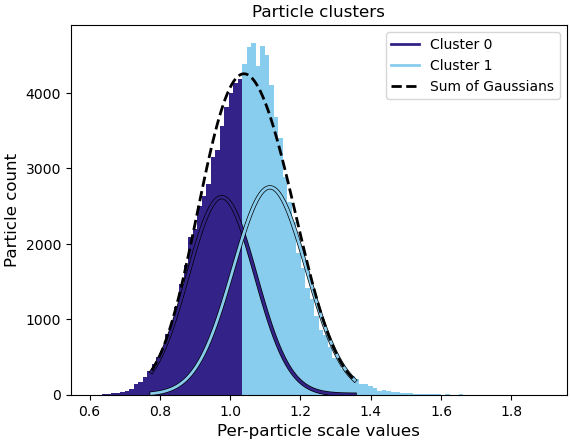
We also tried various NU refinement strategies, including reducing the AWF factor to 1.5, enabling and disabling the dynamic mask (with dynamic start resolution of 1 Å), but none of these approaches significantly improved the map quality. Although we know the overall architecture of the complex, the fit to the density is poor, and even recent map–model building tools such as CryoAtom were not helpful.
We suspect that the issue might be related to flexibility between parts of the complex, but we are unsure how to properly validate this hypothesis. We also tried performing a 3DFlex refinement, but it seems that either the motion was not recognized or we may not have set up the job correctly.
We would be very grateful for any suggestions or ideas on how to improve the quality of our map or further diagnose the problem.
Thank you very much in advance for your help!
Best regards,
Yury Zgadzay