Dear all,
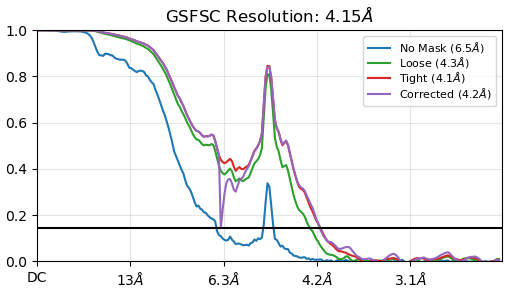
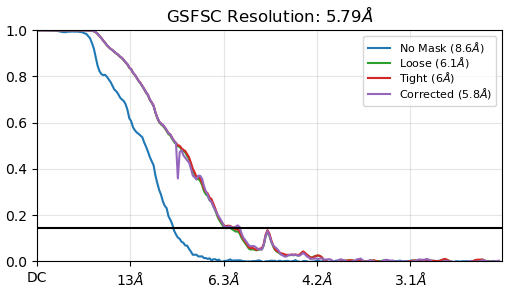
My protein complex is 436kDa, with elongated shape (length around 35nm, width around 6nm), dominant by helices. I extracted particles with box size of 750pxs (pixel size 0.839) and binned to 250pxs, then re-extract with 500pxs (1.5x binned). The homogeneous refinement from 504,796 particles showed a small up-tip within FSC curves near reported resolution. Then a NU refinement job followed (png file). The up-tip got a lot larger after NU refinement. I checked the mask which is quite ok. I also tried different mask thresholds, no improvement. Regarding the 3D volumes, the map from NU refinement got worse than homogenous refinement.
Did anybody ever observe this kind of FSC curves? What is/are the problems?
Thank you.
Min
Hi Min,
Do you see any artefacts in real space? How does the map look? Do you see clear secondary structural features? Also, how do the micrographs look - do you see any unusual/repeated features that could be gain reference related artefacts?
Hi Oli,
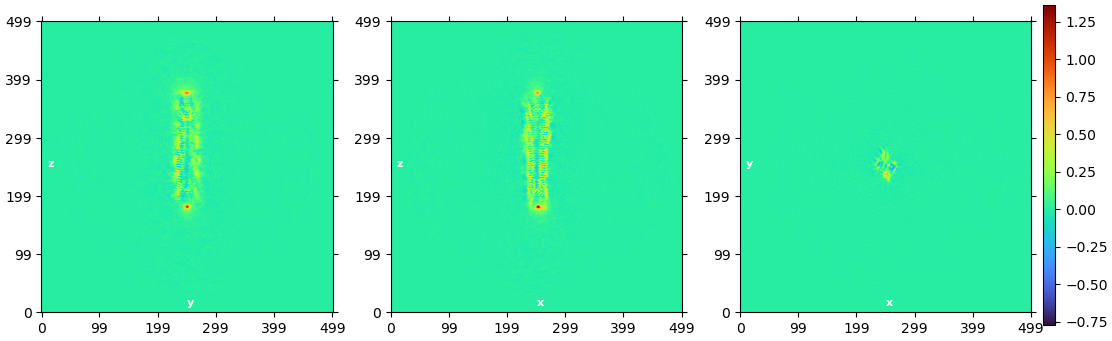
Thanks for your rapid reply. The map from homogeneous refinement looks normal to me, I can see the helices. But the volume output from NU refinement is worse, map got blurry without clear secondary structure features. Here is the real space slice corresponding to above FSC curve from NU refinement
Regarding the micrographs, I did not see clear unusual features. The gain reference was applied before data collection.
Hi Min
You may want to make the dynamic mask loose. Perhaps try changing the dynamic mask far from the default of 14Å to something like 25Å?
Best
Indrajit
Hi Indrajit,
Thank you! Will try and come back to report here.
Hi Min,
What is the initial lowpass you are using in NU-refine, and are you applying any symmetry?
Hi Oli,
Initial lowpass for NU-refinement is 30A (default). I tried to apply symmetry C2, the outputs are very similar to C1.
This is the FSC curves of homogeneous refinement (smaller up-tip):
Hi Min, for something like this with minimal low resolution features and repetitive elements, I would recommend starting at higher resolution - 15 or even 12 Å, assuming your initial ab initio model is good, with clear secondary structural features. This will minimize the chances of persistent misalignment (which I suspect may be the issue here).
Incidentally what parameters are you using for ab initio? For something like this, I would recommend higher res there too - e.g. initial res 9, final res 7, or thereabouts.
Also, it looks like your assembly may have, higher, non-point group symmetry (pseudo-helical)? If this is the case, you may want to exploit it to improve resolution. To do this, see the “custom symmetry expansion” section of this tutorial: Case Study: End-to-end processing of encapsulated ferritin (EMPIAR-10716) | CryoSPARC Guide
1 Like
Hi Oli,
For ab initio, I used all default parameters, only change the number of classes. I tried to increase the initial and final minibatch size, but the outputs were worse. As you suggested, I’ll try to start with higher resolution.
I think the initial model makes sense, comparing with the 2D classes
Indeed, the complex has some repetitive helices. I’ll check the tutorial you mentioned.
1 Like
Good luck! In ab initio, you might also want to test disabling the parameters “center in real space” and “enforce non-negativity”. In certain cases (including helical assemblies), disabling these two parameters can help obtain a model with more isotropic density, which then serves as a better starting point for refinement.
1 Like
Thank you Oli. Ab initio more or less worked. Heterogeneous refinement worked for the first round right after ab initio, when there are still quite some junk particles. When the particles were cleaned a bit, heterogeneous refinement, 3D classification, 3D viability, especially focused classification, all did not work. I always got classes with evenly distributed particles and similar volumes. I relied a lot on 2D classification which is not very reliable.
If anybody has experience on 3D classification on this kind of elongated and helical complex, please share with me. Thanks a lot.
When you say more or less worked, what do you mean? Did you see clean helices, with isotropic density? If not, there is probably more room for improvement.
I would focus on improving your ab initio and consensus refinement first, before proceeding to 3D classification or 3D-VA, as both these approaches are critically dependent on the quality of the input alignments (particle orientations/offsets).
More or less worked means I saw the meaning full architecture that I expected. I cannot see clear helices. At that moment, particles were 3x binned 750pxs–>250pxs
Yellow class has most of good particles, but when I did 2D on the other classes, also a few good 2D can be found. I think some particles were not aligned well. Agree with you.
1 Like
cool - yep definitely would try ab initio at higher resolution (on less binned particles) then! good luck!
Thank you Oli. Will do it and update to you later.
1 Like
What you are seeing is a peak at 5.4A, which is the pitch of an alpha helix.
I have seen this effect with a similarly helical extended protein.
1 Like
Hi Donaldb,
Thanks a lot for the point. Did you do anything to improve it?
Unfortunately I didn’t find a solution to it. My peak was less pronounced and luckily the resolution improved so the FSC was much nearer 1 at 5.4A. The map from NU-refinement did, however, look quite strange in these repeated helical regions.
The main thing I would recommend, as you have already seen is to avoid non-uniform refinement until your resolution improves, as the non-uniform filtering seems to exacerbate this peak.
Thank you Donaldb for sharing your experience. I also learned that NU-refinement should be avoided.