Hi everyone,
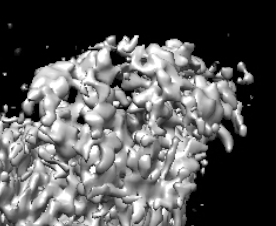
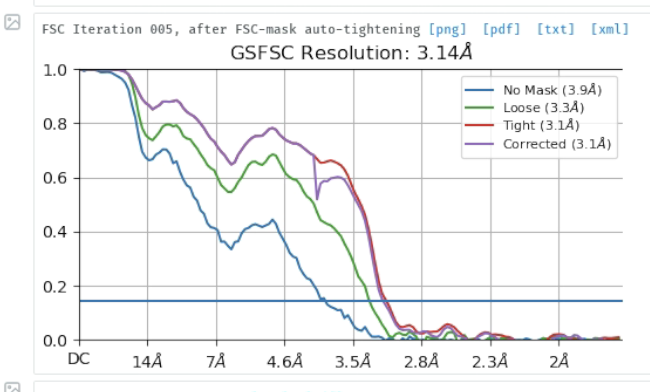
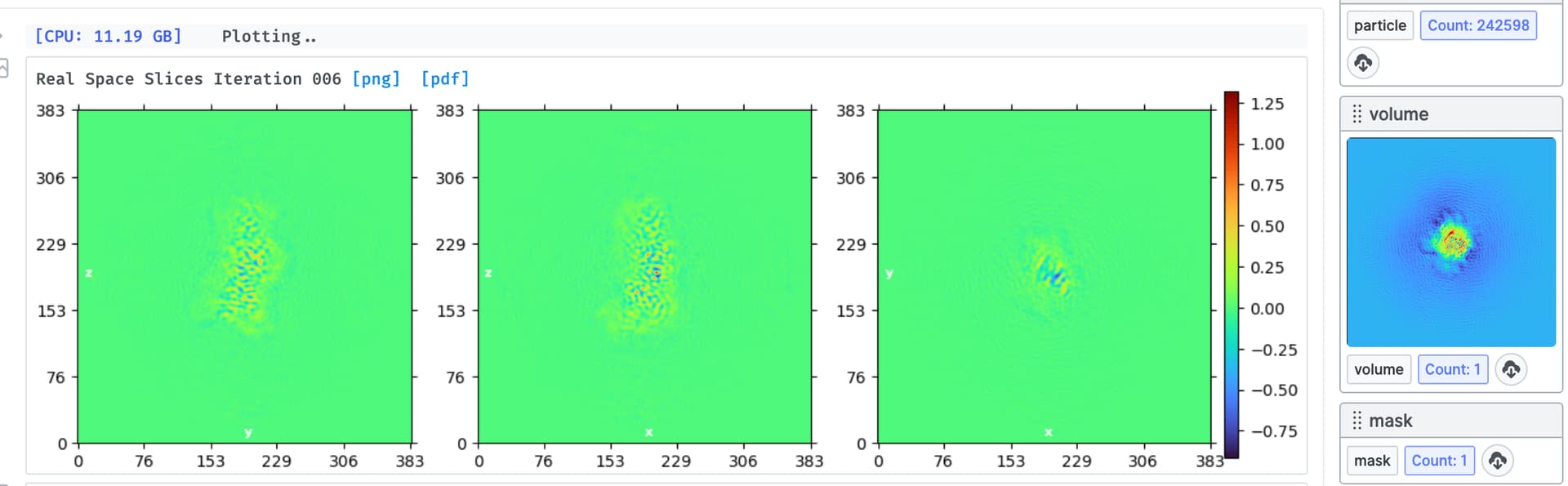
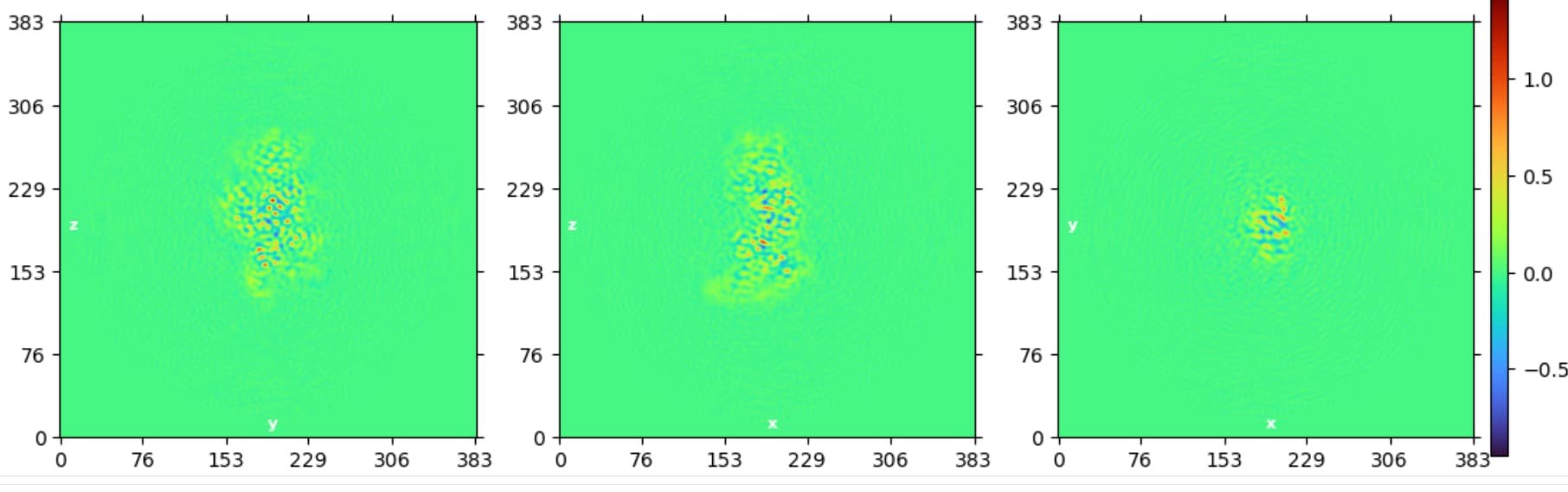
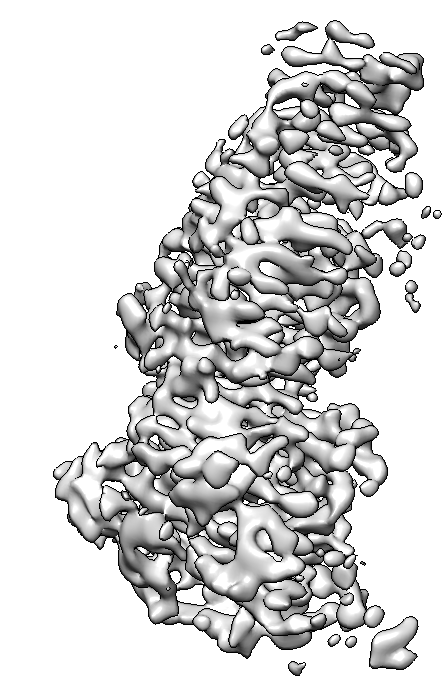
I am trying to process a Cryo-EM data (a small protein (90 Kd) in complex with RNA (30 Kd)), the 2D classification looks great and the NU refinement reports a resolution of 3.1 A, the overall map looks good (I could clearly see the RNA structure) but I found that the map quality is quite bad, the main chain is not connected and lost most details, please see below. It seems like the protein does not have an orientation issue, because we can reconstruct the map from relative low-resolution data and are able to dock the most domains in the map. Someone told me that it’s the refinement alignment issue for this high-resolution data, but could not figure it out. Does anyone have the same expertise, and does anyone know the possible solution? Really appreciate!
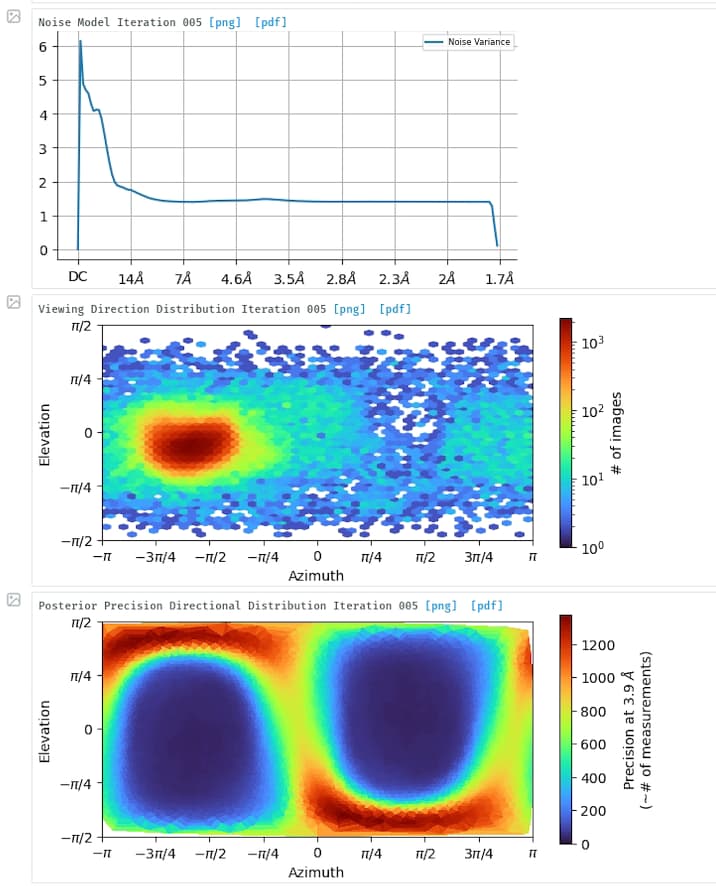
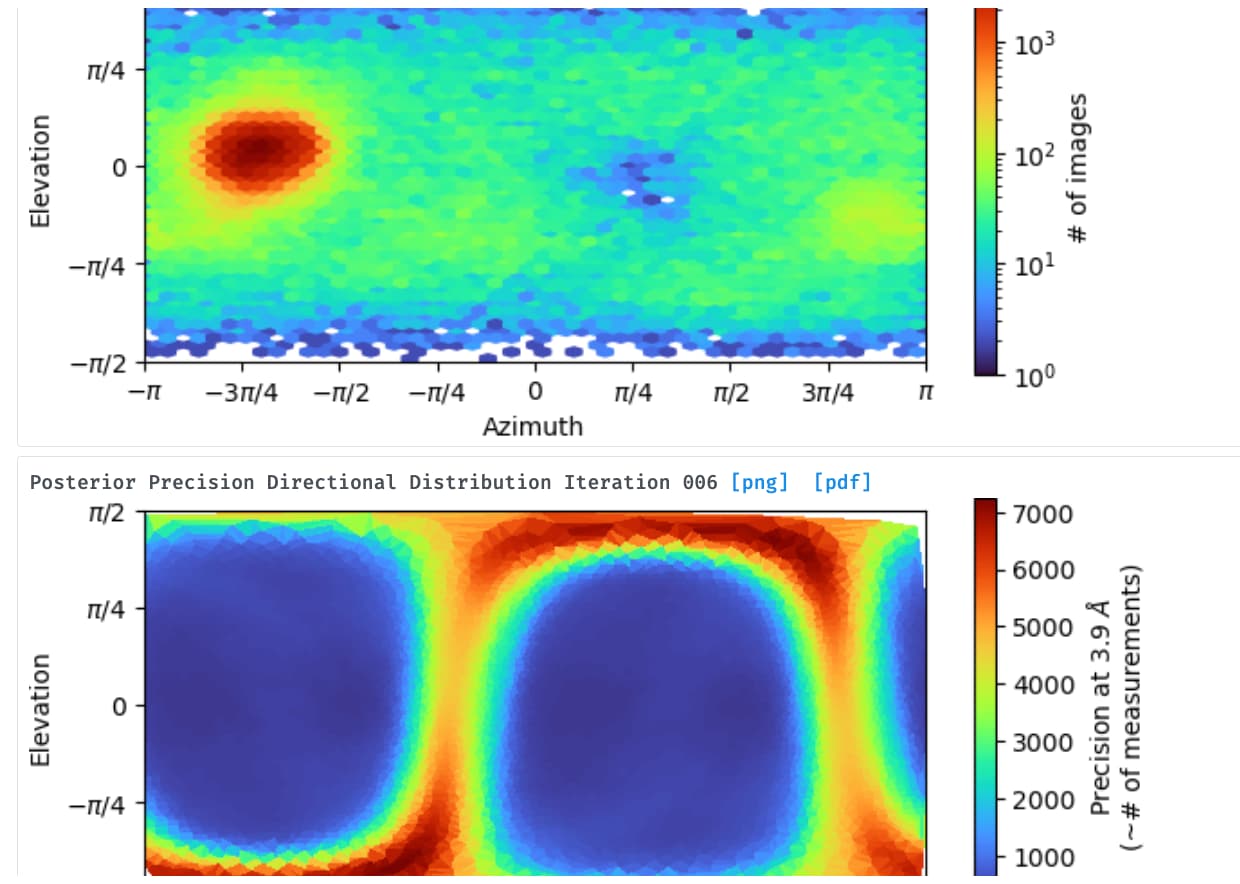
Thank you very much for your suggestions. When I got this map, the first idea is that there must be a preferred orientation. However, we found that the test dataset collected from a 200 kV microscope with the same grads was able to be processed smoothly with no orientation issue, can not figure out what happened when we transfer to collect high-resolution data, maybe we lost important views during collection. I have tried to use the rebalance 2D class in cryoSPARC but didn’t work. I will try to figure out how to balance the views during sample preparation and data collection.
Thank you again!
If of interest, there is a script by Chris Russo called cryoEF that will give suggestions regarding the best tilt angles to fill the missing orientations.
Moreover, if your 90 kDa complex has an elongated form, it is possible that several of the views are just too thin to be properly picked and aligned… if that’s true, then you’d need to make it bigger somehow.
You can also review the process of selection - how did you get to that 150 k ptcls set - to see if the missing views were excluded at some point.
And from that same 150 k ptcls set, what happens if you run 3D classification asking for 4 or 5 classes? (or more if you wish) - the question here is how flexible the complex is.
I disagree that the main issue here is an underrepresentation of 2D projections for some angles. We had various datasets where the angular distribution looked similar to what you show, for particles maybe a bit bigger, and this did not prevent to reach an acceptable density map. When you have a 180° coverage that should work.
I have a few questions that might help you:
How many particles you have after cleaning up your 2D classes ?
How does your ab-initio look like ? Do you have clear secondary structure there ?
What parameters you use for your ab-initio ?
If you have a lot of particles, you need maybe to clean it more. There are various discussions about parameters used for ab-initio that may came at hand for small particles. Have a look at those threads. There are also a couple of threads about parameters for NU-refinement, but you should think about this only when your ab-initio is satisfactory. Cheers
PS: I saw only after you had 150k particles. Conclusion: you need more (high-quality) particles. Collect more data and merge datasets ! Good luck
Thank you very much for your suggestions. We are considering collecting tilted data, but it may not solve the problem with such a small protein.
I have tried to do several rounds of 3D classification with 1-6 classes and 3-6 A resolutions but made no difference, it seems that there is no larger conformation change of the complex.
I am thinking about maybe I can focus on the process of particle selection for this data because I found there is one dominated view of 2D, maybe I need to throw away part of them to balance the views?
Thank you for your good suggestions! After 2D classes, I got about 400k particles, after 3D classes and heterogeneous refinement the number of particles goes to 150-200k, and I could get a good model after ab-initio and the model be much better after heterogeneous refinement. I believed the biggest issue for this data is the 3D refinement not working, I will definitely try more parameters and methods to do the 3D refinement.
Thank you!
I only mentionned CryoEF because I’m facing a similar problem, and you are right, from what people say, tilted data adds some complications, and it is not 100% sure to work in both our cases.
Thank you for bringing this up to discussion, I’ll keep following the topic.
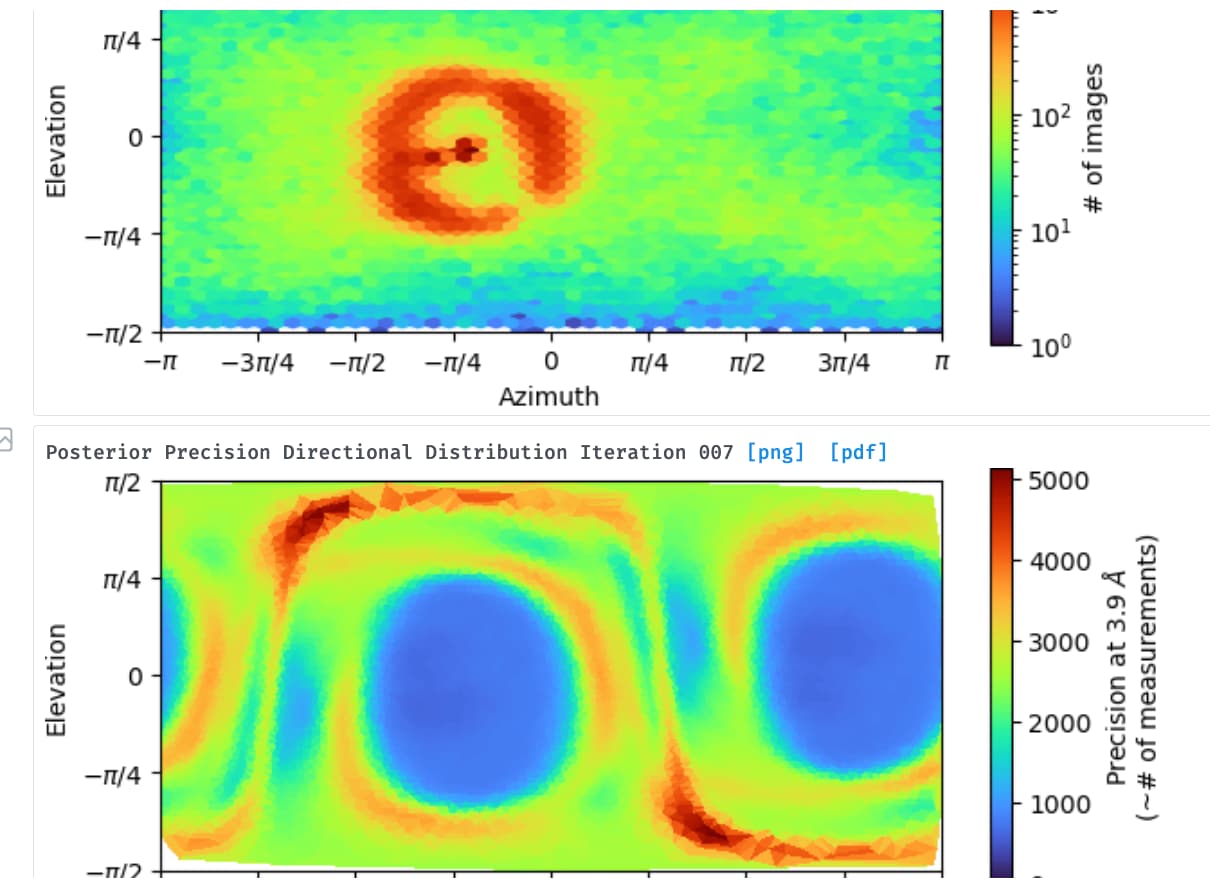
My protein is very similar to yours (104 kD, elongated shape) and I got very similar result as what you have seen. But in my case the main issue is preferred orientation, without tilt i saw below
I agree with @carlos that “it is possible that several of the views are just too thin to be properly picked and aligned…”. but it is tricky to make it bigger and rigid.
I tried cryoEF before, it shows recommended tilt angle is 23-38 degree, we collected 40 degree recently and now all my particles cannot get aligned
Would love to see whether you have made new progress and discuss together for solving challenging small elongated particles.
Your reply is so helpful! I think i have the same problem for small protein. Some views are just too hard to get aligned. I was told by several people to skip 2d and go directly to 3D but at least in cryosparc i have not seem much improvement. Do you run 3D classification in cryosparc or relion? Any specific parameter recommended?
Thanks a lot!
What we do is to go straight to heterogeneous refinements using a good map as first, then bogus maps as 2nd, 3rd, 4th… (I did not invent this, it’s somewhere in this forum), then keep going with heterogeneous refinements until a good set is selected. This helps.
I still do 2D classifications just to have a look at the particle set, and sometimes eliminate clearly junk classes.
Sorry I still did not get out of it, so I hope it’ll help you…
Yes, I believe we got very similar results, unfortunately, I still work on it and could not figure it out. We think it is possibly an orientation issue by carefully checking the 2D classification results. We are currently trying to test with different types of grads and different detergents. I will update my progress if I finally work it out.
Thank you! It’s helpful. I did similar thing but usually end up with very similar classes and similar amount of particles in each class when try the second or third round of heterogeneous refinement. It is so hard to get clear junk after first round of heterogeneous refinement somehow.
I am curious about the percentage of good class you get from 2D (And do you get any end views for elongated particles?) Mine usually only 20-30% are good (side view only) but for most tutorial data at least 60-80% particles exist in good class with clear secondary structure. So my assumption is I might have 40-60% good particles that could not get aligned somehow due to low signal for alignment. If that’s the case only increase the particle size will help…
Looking forward to your update. Hope people who see this and has the same issue can update and discuss about the possible solutions here.
I am not sure about the percentage of the good 2D classes since I have not gotten a good high-resolution map. By checking the 2D classification, there are maybe some very small parts of end views like sphere shape not elongate shape, but I don’t know whether this view could help alignment, the resolution of sphere shape views are quite low and without secondary structures. We are focusing on how to get another small elongated view.