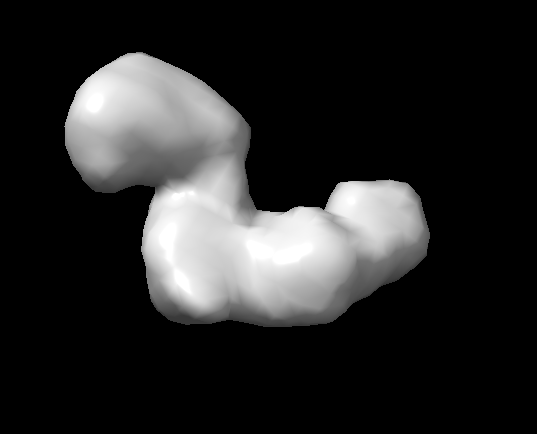
using negstain-data, i got this ab-initial map
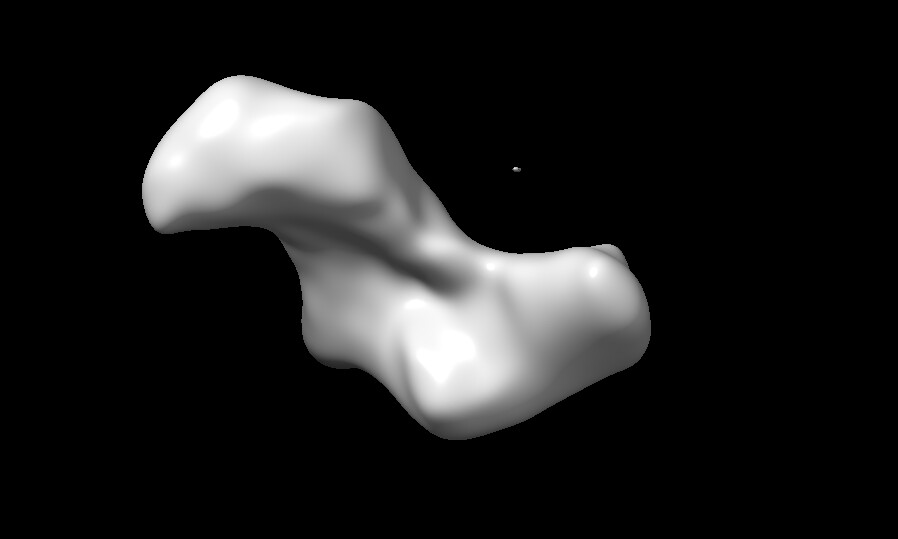
but after i do NU-refine (all using default-settings), it got worse. can i repair this problem? or maybe it won’t get any better because of my sample property?
Really thanks a lot!
using negstain-data, i got this ab-initial map
but after i do NU-refine (all using default-settings), it got worse. can i repair this problem? or maybe it won’t get any better because of my sample property?
Really thanks a lot!
How large is this particle? Looks rather like you’ve hit the resolution limit of negative stain data, which is ~12Ang (if optimistic) and ~20Ang (if pessimistic). ![]()
my particle is about 100A or more, ab-ini is reported to be12.63A.
in this situation, do I need to do nu-refine anymore? my teacher let me finish this process, yet don’t know why after NU-refine, it looks worse
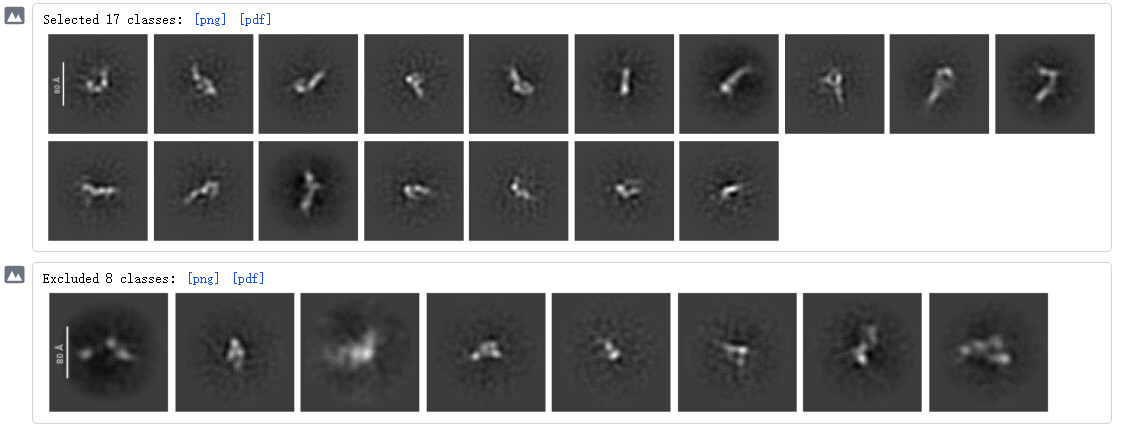
Would you share a screenshot of your 2D classes?
Thanks. ![]()
It looks like you’ve not got many particles. Chances are you’ve hit the limit of the data you have in your hands… especially if it’s neg stain, which imposes a resolution limit impossible to overcome in neg stain.
if i make new sample, may I prove it in NU-refine?
or maybe should i stop here, the ab-ini is pretty much enough ![]()
If you move to cryo grids, you’ll likely be able to improve your sample - but cryo is a very different world to negative stain!
Thanks a lot, I’ll try it later ![]()
Agree with @rbs_sci . On NS it is not worth collecting too many images. With 10 k ptcls or less, you’ll reach the limits of what one can do with NS. You’ll never see secondary structure because of the dye coating. If you have a lot of particles, you can try multi-class ab-initio reconstructions to see if it can show you some different conformations - just to have that information, which might be useful. I don’t think it’s worth trying to refine anything, though (check your initial FFTs, how many rings do you see? I bet not many). If you still want to try refining for fun, set max resolution to 20 angstroms or so. But don’t expect miracles.
Oh, by the way, looking at your 2D classes makes me think it is too soon to go for cryoEM. I’d rather try to stabilize the protein somehow (mutation(s), partner(s), antibody, some ligand that makes it more rigid…). You can use NS to optimise that.