Hi,
Do I need to run a homogeneous refinement after selecting particles from cluster 3DVA display mode for a second round of 3DVA or can I use the output particles and volume directly?
Best regards,
Nuno
Hi,
Do I need to run a homogeneous refinement after selecting particles from cluster 3DVA display mode for a second round of 3DVA or can I use the output particles and volume directly?
Best regards,
Nuno
Depends on what you want to achieve but generally speaking, yes, you can use the particles and the volume directly for refinement. Do make sure that your particles used for refinement are not downsampled. Generally 3DVA runs much faster using adequately downsampled particles.
Hi Nuno,
I don’t think you need to run a refinement (for alignement) in-between two rounds of 3DVA, but I usually do it, believing that this new round of refinement will be more efficient than the first one.
Thanks for the replies!
Best,
Nuno
Hi all! Just a quick addendum to the excellent replies:
3DVA assumes that all particle alignments are fixed. Depending on the type of heterogeneity present and cluster size, it can be useful to do a refinement for a particular cluster to improve the alignment quality. However, this may not always be the case (e.g., the cluster may have too few particles which may lead to worse alignments than the original consensus refinement).
Out of curiosity, @nbustorff, can you say a bit more about your multiple-round 3DVA approach? Are you running 3DVA again on each one of the clusters? Have you found this to be a useful strategy to separate classes further?
Thanks!
Valentin
Dear @vperetroukhin,
I have been using exactly this strategy. It is very useful to further separate classes. Although, I recently became unsure about the true meaning of the clusters. What I mean by this is the following.
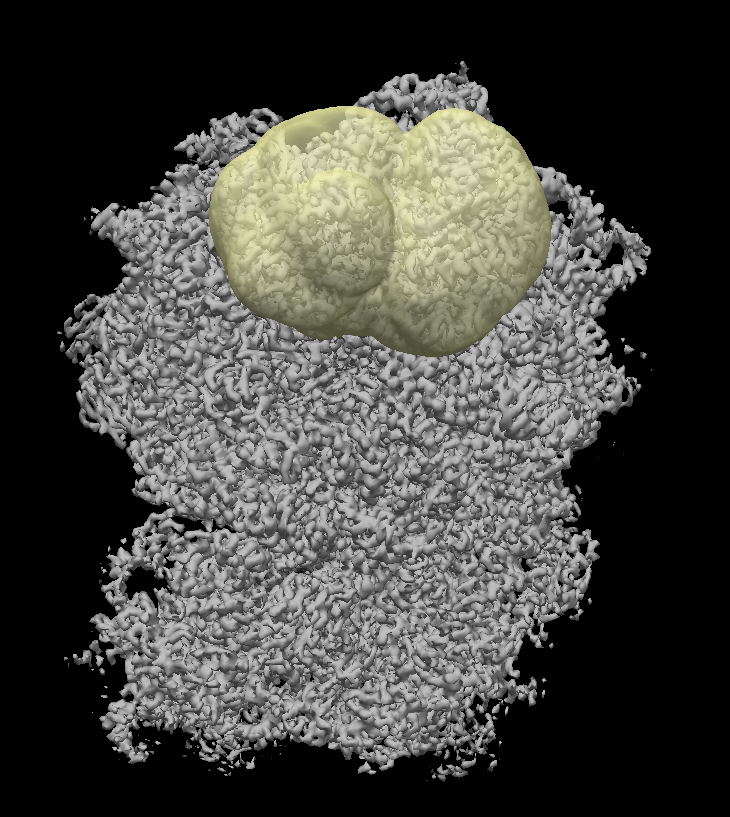
Although the job seems to work. For some masks, the 3D map from the reconstructions look artificial in the mask region. They look like they have mask induced artifacts. I am a little puzzled.
I am working with translation of ribosomes. One of the many things I tried was to cluster particles that have the tRNA occupied differently in the PTC. Either they have A/P/E sites occupied.
I am working with downsampled 256px particles because our computers can’t handle full particles of 512px.
The way I create the mask is:
Chimera
Cryosparc
input: map
output: maks
threshold: depending on the molnmap volume (normally: 0.1)
fill holes: true
dilation: 3
padding: 6
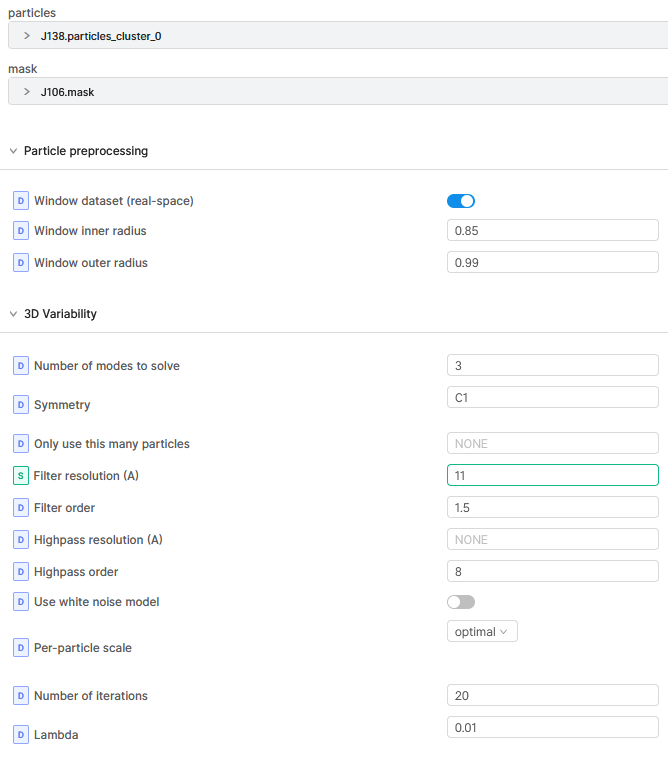
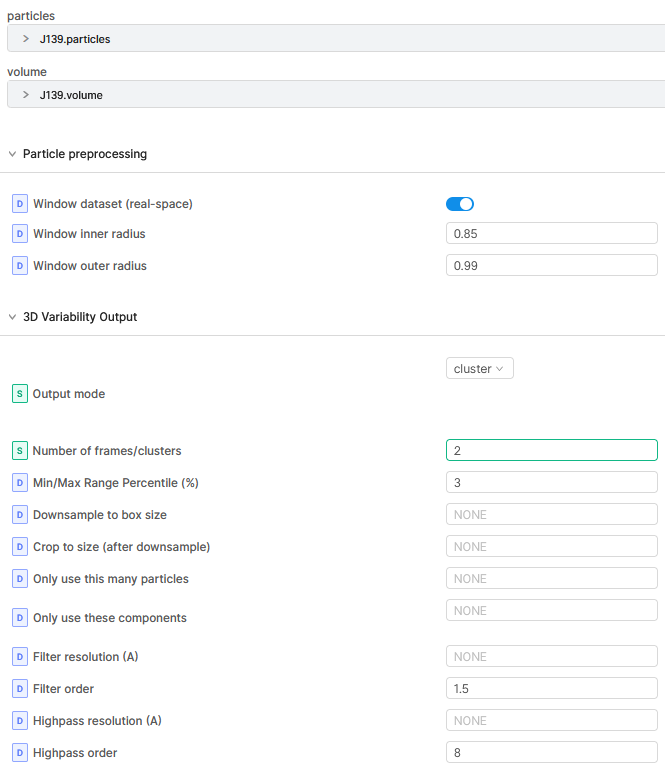
Mostly I run 3DVA with 3 modes. Depending on the result components graphs I choose the number of clusters for the 3DVA display job.
I am super happy with the 3DVA! I would like to understand it a bit better to make sure everything is working as it should and that I am not introducing artifacts.
Thanks a lot.
Best,
Nuno
Thanks for the description!
Although the job seems to work. For some masks, the 3D map from the reconstructions look artificial in the mask region. They look like they have mask induced artifacts. I am a little puzzled.
Ah, strange. Could you share a screenshot of these artifacts?
Dear @vperetroukhin,
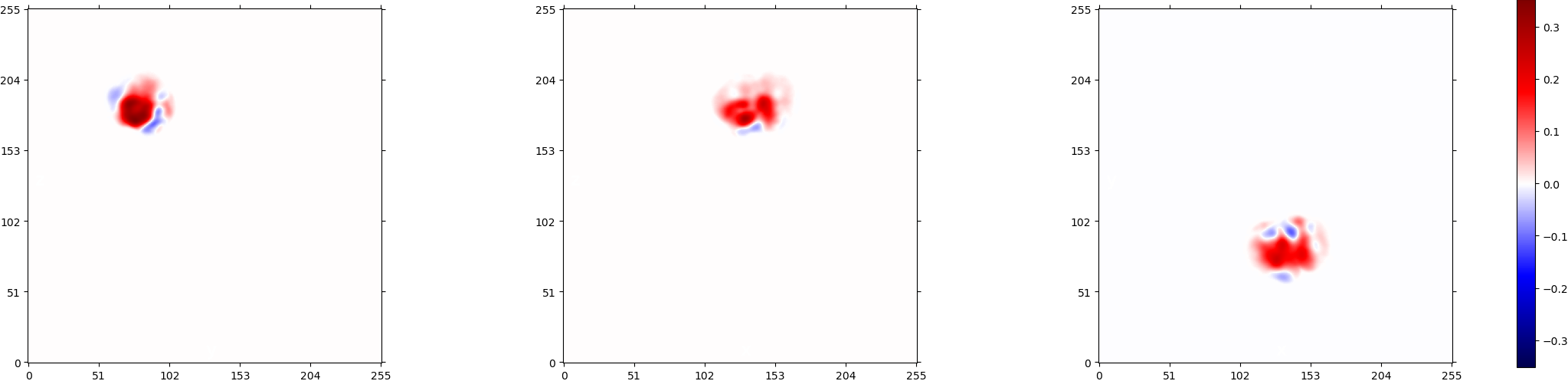
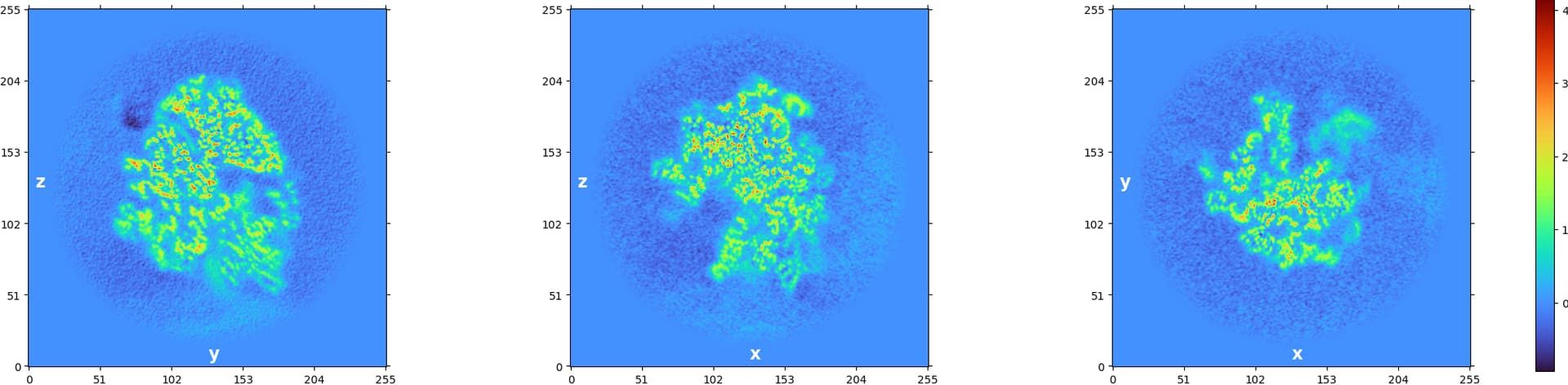
I hope these figures can help. I wish that the density is real, but as you can see in figure 10, it is not very convincing.
Best,
Nuno
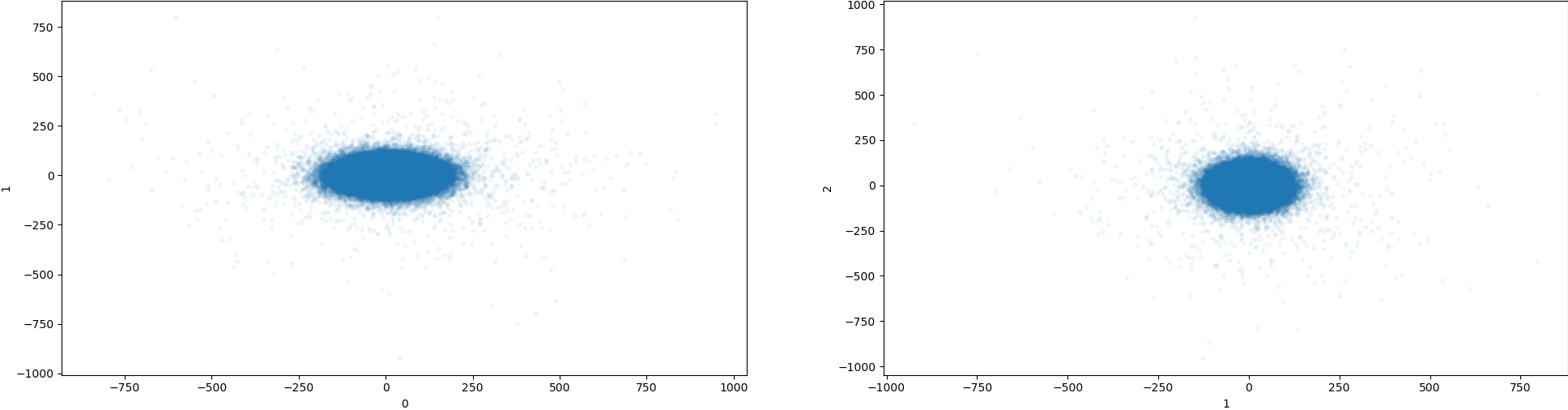
3DVA Job:
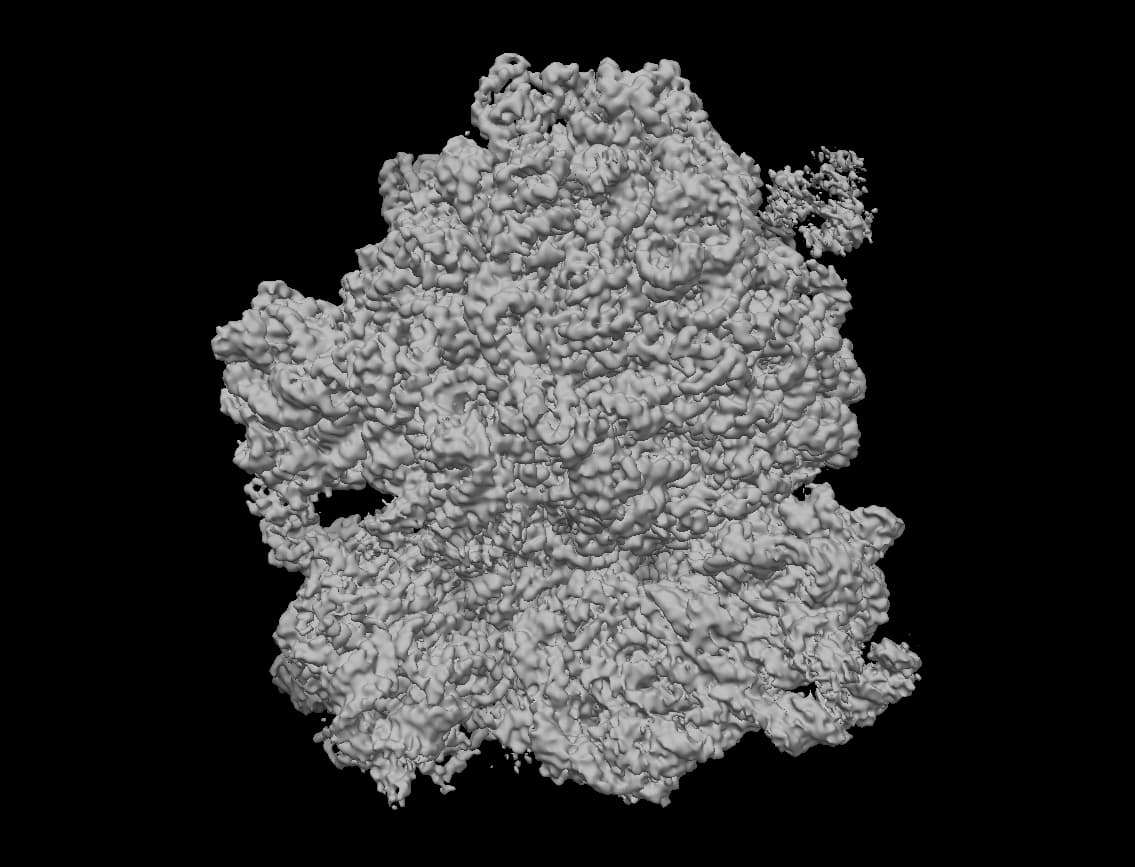
3DVA display Job:
Ah interesting, thanks for the figures (and the numbering  !). It is quite difficult to tell if that’s real density, especially because of the relatively large negative values you see in Figure 9 . A couple suggestions:
!). It is quite difficult to tell if that’s real density, especially because of the relatively large negative values you see in Figure 9 . A couple suggestions:
Thanks for the suggestions. I will definitely try.
Best,
Nuno