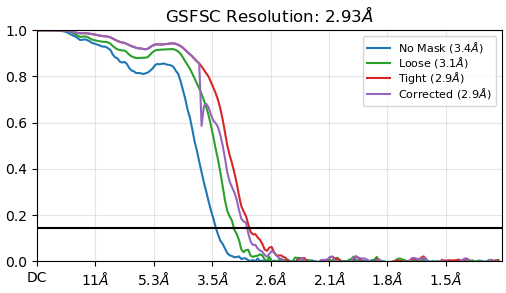
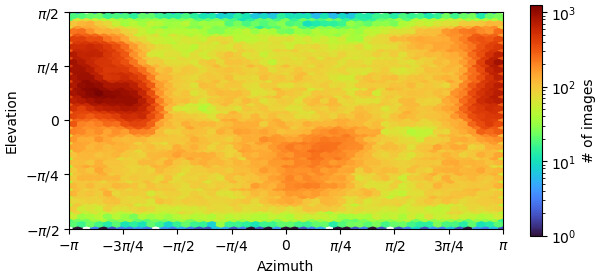
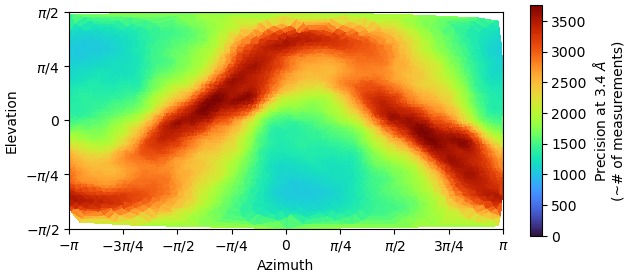
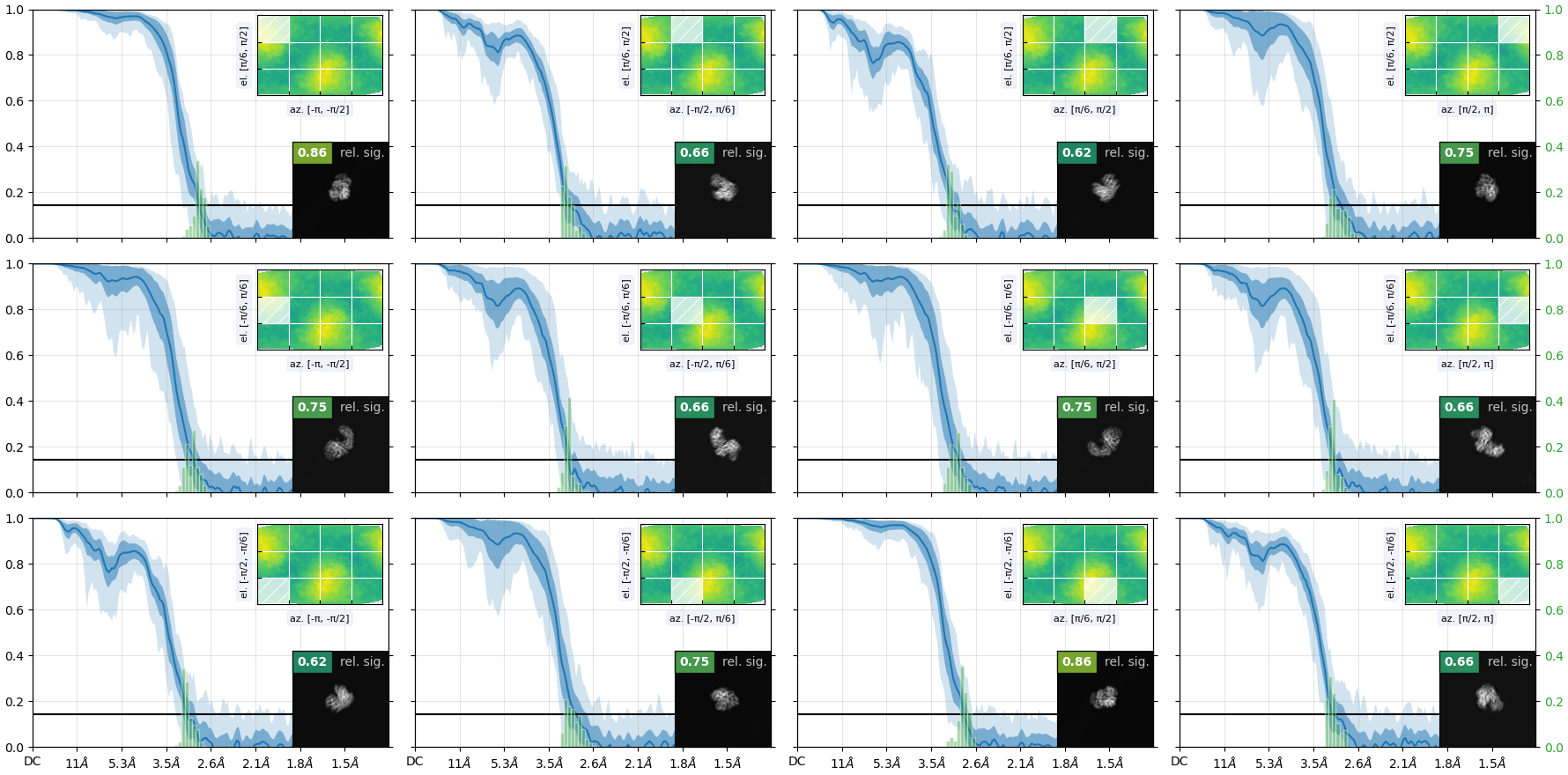
my reconstruction is probably suffering from missing views or misalignment. I post some images below of the final GSFSC curves and a 2D classification of the full particle set (~400000 particles). No matter how much I clean or classify this dataset in 2D/3D, I always end up with the same orientation problem and map quality that prevents me to properly build a structure. I suspect that I miss some views along the particle long axis but I would like to be sure.
So, starting from the 3D reconstruction, how do I figure out which 2D classes are missing or which are the less represented ones ? I plan to train Topaz with those classes and find the missing particles (if they exist in my dataset!)
Is there a general consensus method for doing this ?
I’ve seen much worse in terms of preferred orientations. What makes you think that is the problem? Does your map look too flat or too distorted in one direction? You can try rebalancing the particles using the 3D error criteria to eliminate the worst ones - you seem to have enough particles for that. If you don’t see internal features in the map, but it does seem to have the expected shape to some extent, you can try 3DVA on the entire set to try to understand the heterogeneity. - Waiting to see what others have to say.
density is stronger in one direction and weaker in another direction, and some alpha helices lose the continuity in the main chain. unfortunately, what I need to see is in that portion of the map. i’ve tried rebalance 2D/3D too but the effect is still present and I lose resolution.
so, that’s why I would like to find out which views are actually missing and train a Topaz to specifically pick them. What I am looking for is a graph or an image that tells me the contribution of each orientation/class to the final resolution, in order to pick the weakest ones and train topaz… is there something like this in Cryosparc ?
I agree that things don’t look too bad here, but to answer your original question, you could try:
(1) Running the Orientation Diagnostics job. Unfortunately I think there isn’t a way to make it give you more than 12 projections in the “Avg. Relative Signam Amount within Azimuth-Elevation Viewing Regions”, but it does give you a general idea of which projections correspond to which parts of the orientation graph and what the relative signal is, which I find helpful. Plus it gives you the Relion-esque .bild image above this of which directions have more and less signal, which is a little more intuitive for me than just the classic image you show.
(2) You can play around a bit with Create Templates and/or Reference Based Auto Select 2D using “cluster” as the selection mode. The former will give you 2Ds that you can compare to your 2Ds and see if much is missing, and the latter will output a side-by-side of your 2D classes and the 2D classes that may give you something interesting, or may not. It is not formatted for this use (see Allow interactive editing of reference based select 2D? - #7 by olibclarke ; you’re not alone for wanting this), but poking around may give you something.
Edited to add: if your picking is good and your particles are clear in your initial micrographs, it is likely not a picking problem (could be a preferred orientation problem on the grids, or could be some other issue like flexibility etc). I find a lot of users don’t spend enough time looking at their micrographs and their initial picking and then spend a lot of time optimizing picking when in reality those views are just not there. For good micrographs blob picking with some work with the sliders in Inspect Picks does a great job, although Topaz is good for more complex situations, which yours may fall into. If you have everything else great on the micrographs and you think that the preferred orientation is really what is limiting you, try taking a tilted data set and see if the issue resolves.
this reconstruction comes from 3 grids of the same protein, 28000 movies, so quite a lot of data.
we started data processing by separately autopicking a small subset of each grid, then trained topaz to pick everything. after several rounds of 2D/3D classification this is the best results I have. I think the main problem comes from the shape of the particle (elongated) with no symmetry, small, and flexible too (what we see in the map corresponds to 100KDa). There are a few projections (mainly the ones along the longest axis of the particles) that are really small and probably difficult to align. But I will keep trying…
I found out the image you are talking about (thank you!!!): clearly some projections give less signal than others. I can try to manually find those views and train a topaz, or do a Reference Picking (but I know already this is not working well my protein), then merge everything and remove the duplicates, to see if I can get more views. But sounds like a huge trial-and-error thing, and I was looking to a more “rational” approach… if actually exists…
Those look relatively good still – I think the colors get red on the relative signal when it gets close to zero, and the FSCs + histograms look pretty consistent throughout orientations. You can take a look at your cFAR value to see if there is a likelihood that it is causing the map issues you are seeing; if your value is over 0.8 or so you’re probably good (but I have also seen maps with much lower cFAR values that are fine for what is needed). Take a look at the orientation diagnostics ( Tutorial: Orientation Diagnostics | CryoSPARC Guide ) guide for an example of more pathological preferred orientation.
You could also try generating a certain number of templates with Create Templates, then do a Rebalance 2D Classes job and take a look at the event log at the number of particles assigned to each 2D class that you generated (just do the same number of superclasses as templates you generated). That will let you match which classes have fewer assigned particles, although it will be somewhat manual. I would be surprised if preferred orientation is causing your underlying issue here. But I am often surprised
Other thoughts:
-Are you seeing the stretching in the sharpened and the unsharpened map? Have you played around with different b-factors in postprocessing? That can cause similar stretching/streaking in some maps. You can also try deepEMHancer, local b-factor sharpening (third-party, such as LocSpiral), or density modification (Phenix) and see if any of those help with your broader map issues.
-Some folks have success in doing a local refinement job where they give the mask of the entire protein for small proteins; people also have luck in changing some of the default parameters in classification to classify better and there are lots of good threads on the forum for small protein processing and parameters to use.
-If you are trying to actually use the rebalance job, I would definitely get rid of particles on the basis of alignment errors so that the particles with the most solid alignments remain. But again you will usually lose resolution if you get rid of particles, even overrepresented ones.
-Have you completed reference-based motion correction? This is very helpful to improve resolution over a small threshold. Also depending on what grids this data set was collected on, gold/gold grids often give much better data than carbon, particularly for smaller particles.
The more I look at this, the more it makes me think of a flexibility issue. The C-shape would be opening and closing to some extent, and without looking at the map, my guess is that the helices that loose continuity are (or close to) the fulcrum of that movement. Helices are not that rigid, each turn can have some freedom to shrink and expand depending on the scenario. My first shot would be: 1) mask the biggest side of the C and do a local refinement (expecting the density for the other side to fade out); 2) make a very broad mask around the now invisible opposite side (trying to include all possible positions) and run 3DVA cutting at 8 angstroms or more, you don’t want high resolution at this point. Look at ~10-20 Clusters and Intermediate modes, it should show the C opening and closing, the helices should be continuous in each map; 3) See if you can group clusters that are close to each other to run NU-local refinements and improve resolution of the helices.
BTW, have you made a movie with the results from Rebalance 2D classes job on the side views? Don’t you see this movement? Even if it doesn’t look too dramatic, if the helices are the fulcrum, they will be hard to align.
to reply to everybody: there is for sure flexibility, everything is moving in this protein! already, by masking one small domain and classifying I obtained a better set of particles and better maps although the orientation distribution is the same. I think there is a combination of everything, small particle, flexible, probably some preferential orientation, combined with my limited experience ! I will carefully read and try all your suggestions and let you know the result. thanks a lot !