I am struggling with a membrane protein of Mw 100 kDa in detergent. I could see good 2D classes in the top and side view. Moreover, after reconstruction (ab-intio, hetero-refine and Nu-refine), we are never been able to get high-resolution maps of the transmembrane domain.
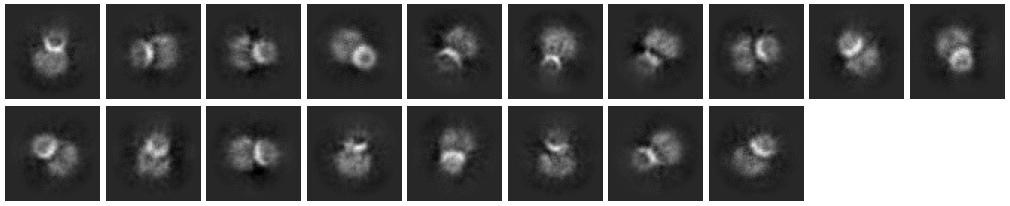
I have labeled 8 class which I guess it is a side view. In fact, this protein has a big intracellular domain and a transmembrane domain. In the begaining, after 2D classification, this protein was found to have an orientation advantage (almost top view). So, I added detergent to eliminate orientation.
When I select all top view to further refine (ab-initio and NU-refine), I can not find transmembrane domain in the map. So, I chioce side view particles stacks in the 2D class (e.g., figure below) to run further refine.
But the map is still bad.
Is it possible that the detergent is wrapping the transmembrane domain of my protein so I can not get a high resolution map of that region? and how can I fix this proble?
Thanks!
wu
These do not look like side views to me I’m afraid - they look like mis-centered picks where you have two particles close together. I would consider the possibility that the detergent has not worked yet to eliminate the orientation bias - often we need to screen multiple detergents, or use a substrate like graphene, in order to find something that works for a problem case. You could also try extensively sub-classifying the non-top views, to see if something more obvious pops up?
I would also try running ab initio just with the high res top views - the results will be very stretched, but perhaps will give you some idea what to look for if there are a few side view mixed in there - worth a try!
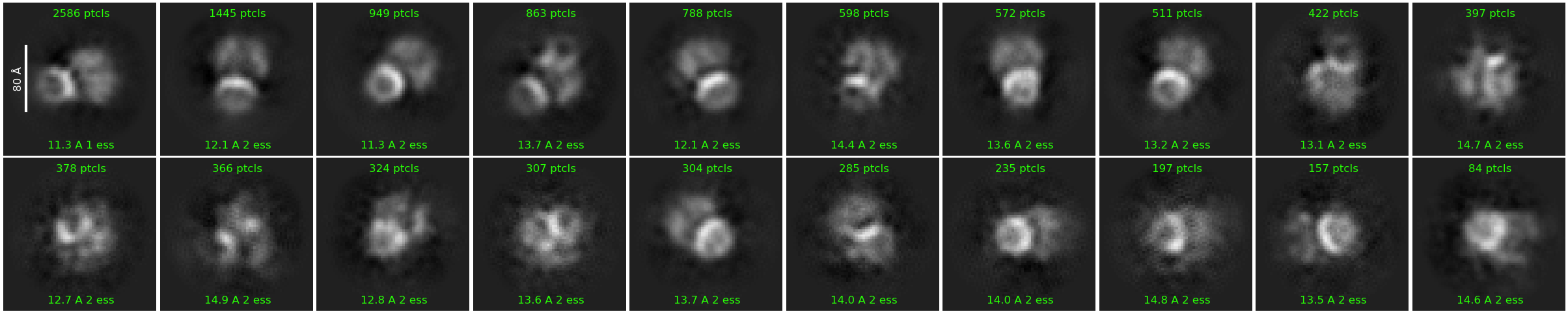
Nearly 20,000 particles (non-top view parcticles represent a small percentage in total)
As you advice, I run the ab-initio for the high res of top views and I will post it here as soon as the results are come in.
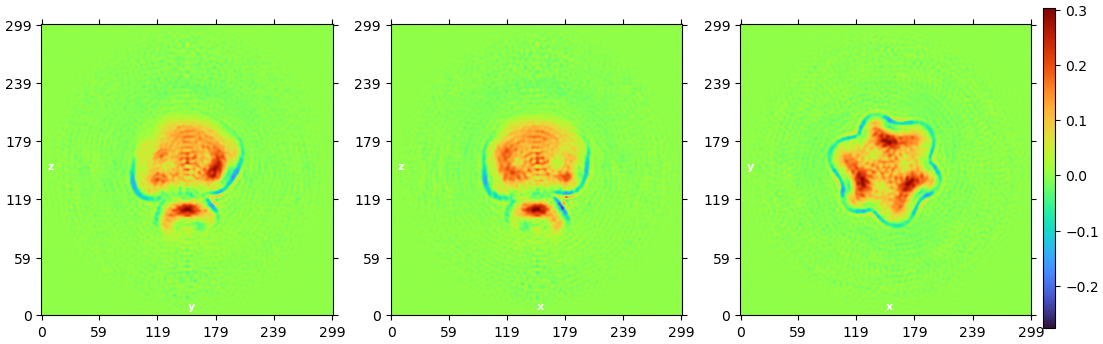
By the way, I have a reference map from other’s published article with a large micelle belt between intracellular and transmembrane domain, I load this map in cryoSPARC and create template, but it has a large difference from my non-top view (below fig). Can this be a evidence that this non-top view is not a atual side view?
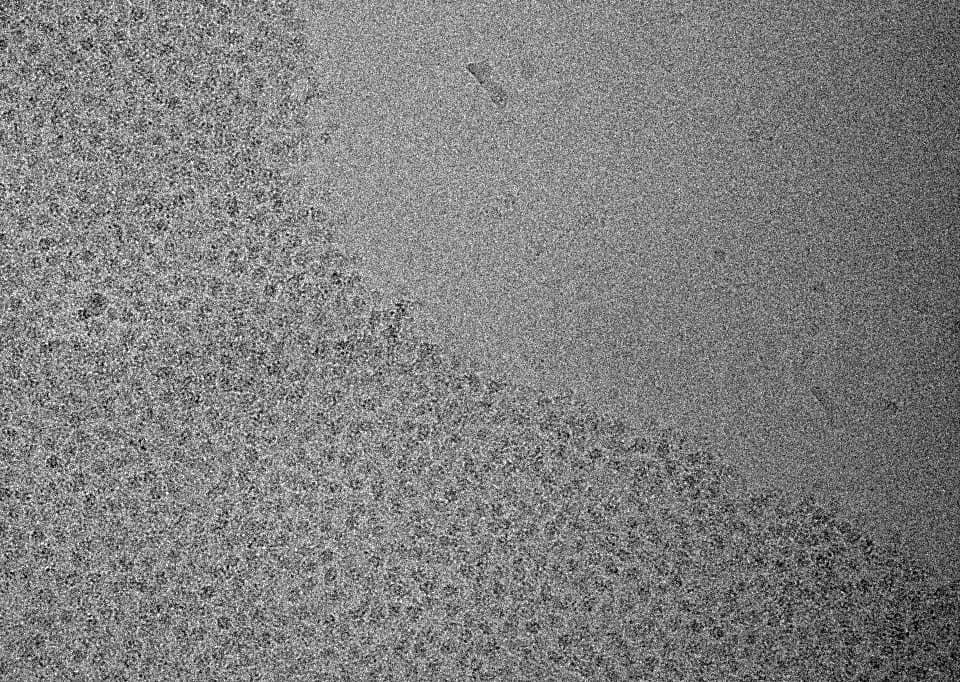
It can. Your sample is far too concentrated - it is clearly overlapping where protein is present on your micrograph and the 2D confirms this with how the classes look.
In your original post you’ve got some nice looking top views…
If Topaz doesn’t work to find any, you could try picking with only a side view (from the templates you created from that EMDB map) and try cleaning it up that way; that sometimes works (leave the low pass filter at default, no matter how tempting it is to try picking higher frequencies!)
But to be honest, when I see micrographs that crowded, I usually go back to playing with grid conditions as it’s often more successful than trying to fight with the data.
I found it hard to believe that this is not what I wanted, because it really does look like a side view (through it only few thousand)
As you advice, how can I use just side view as template (create template will create 50 equally-spaced templates), how can I use part of them to do template picking.