Hi @emil ,
I have a similar protein to yours (~140kDa monomeric, non-symmetric, ~10 N-glycans). My 2D classes showed very clear secondary structure details and fuzzy density on the surface due to glycans. Getting a decent 3D structure has proven to be very difficult for a small protein with such high glycosylation. I had to be sure that all junk was removed and that all particles were monomeric - 3D initial model generation otherwise just gave a smear. I still have some internal heterogeneity (hinging and glycan motion) but have now managed to get a moderate resolution 3D structure with better resolution at the protein core (really looks like ~4/5A).
Glycans are very flexible and they may influence the alignment of your particles in 3D. In my case, the glycan motion occurred along with protein motion so that there was a shell of density on the surface, although resolution reported with FSC curves wasn’t bad. What worked for me was firstly to extend the dynamic mask radius. I have branched complex-type N-glycans and the default of 6A near and 14A far was too tight and parts of the density was cut away (that’s what I think anyway Local B-factor sharpening - Post Processing - cryoSPARC Discuss). Extending the radius to 12A near and 20A far seems better and gave glycan stumps on the surface of lower resolution than the protein core.
To improve the resolution beyond what I get with a consensus refine of all the good 2D classes’ particles, I did ab initio with 4 classes followed by heterogeneous refinement. This gave an improvement which was then even better with non-uniform refine. I only see good glycan density when the resolution overall is rather low. With sharpening they are basically lost.
Here is a pic after NU refine at low and high thresholds showing glycans before sharpening and core density after sharpening.

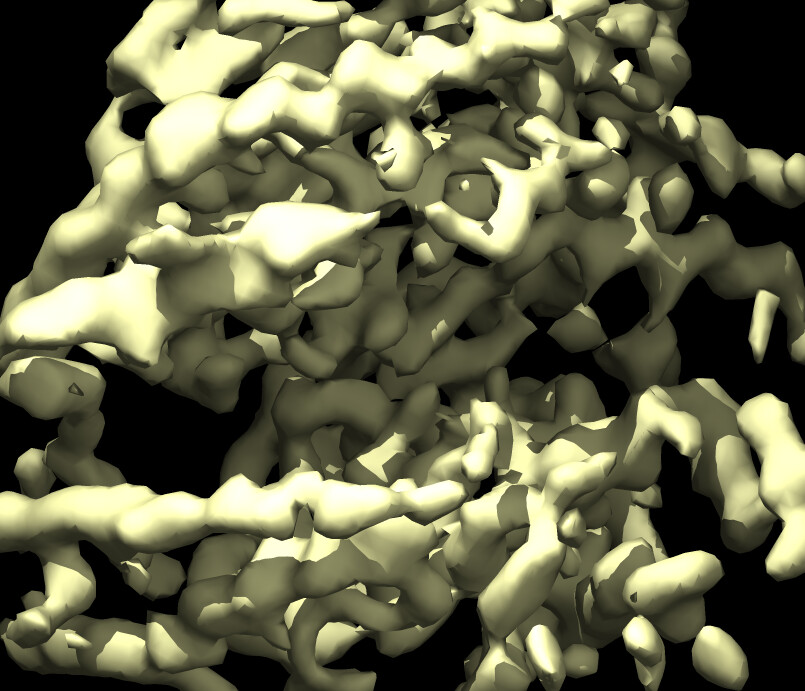
Here is a pic of the output from heterogenous refinement where glycan stumps are seen more clearly

Hoping that this may give you some tips - although I am still in need of some myself. I thought that as long as I get rid of as much protein heterogeneity as possible, glycan heterogeneity would not influence the 3D alignment as much. Perhaps we can get some information about the glycans at the end by using per-pixel B-factor sharpening or something like that? I am trying to use 3D variability analysis to further ‘purify’ my current classes so that I can reconstruct the most homogenous set of particle conformations but not sure how to focus the analysis on protein variability and not glycan variability (Masking glycans for 3D variability analysis - 3D Variability Analysis - cryoSPARC Discuss) Maybe something like that could help you as well?