I am new in the Cryo-EM field. I am trying to solve a 140kDa structure, actual size of my protein is ~30 kDa connected to MBP (~40kDa) which gives ~70kDa protein, overall assembly makes dimer. I tried to refine in NU-refinement (default values), which was giving me 5 A resolution after three iteration but in the final iteration it jumps to 8A. Can we stop it at 5 A or it is a noise?
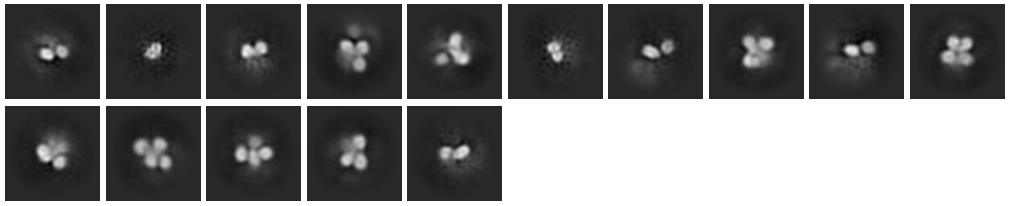
Here I am attaching Select 2D classes Image (Selected and excluded). So this protein make dimer and tetramer. I am able shortlist dimer from tetramer at the time of Template picker using 150 diameter. After that multiple round of 2D classification and finally I am able to select 15 classes out of 50 classes. Which gives me ~43000 particles.
These 2Ds don’t show much evidence for clear secondary structure - if I had to guess I would say that it is likely that the connection between MBP and your protein is a bit too flexible for high resolution reconstruction (at least without a much larger dataset).
Can you fit the MBP unambiguously into your map? At 8Å, you should be able to clearly see helices, and confidently fit known structures.
I also got one map with resolution of 6.7 A, map looks good, problem is all four blobs look similar to me and I do not know which blob should I choose for MBP fitting. Is there any program which can guide me to choose the blob for MBP.
Can you see helices in the density? If not, then it is unlikely to be a “real” 6.7Å. If you can, you should be able to recognize the shape of MBP, and fit it based on that recognition.
I agree with what @olibclarke is saying. Additionally, I would try to clean up your 2Ds even more. I’ve found that experimenting with 2D parameters can really help in certain cases like this. Here are the values I normally change:
-Re-center mask threshold (I increase this to 0.4)
-# of final full iterations (I typically set this to 2)
-#of online-EM iterations (I set this to 40)
-batchsize per class (I set this to 1000)
I had a protein with a flexible component, and while I never got to super high resolution for that particular dataset, I was able to get it close to ~4-5Å, which was enough to help out with modeling and gave us some very interesting insights into the complex of interest. I would also considering taking another dataset–43k particles don’t really give you a lot of room for cleaning and experimenting. For my flexible protein, I had ~15-20k images, and at the end, I only had ~80k particles in each dataset (the particle distribution wasn’t fantastic, and with the flexibility of the system, I had to discard like 80-90% of the particles that were picked). Good luck!