Hi,
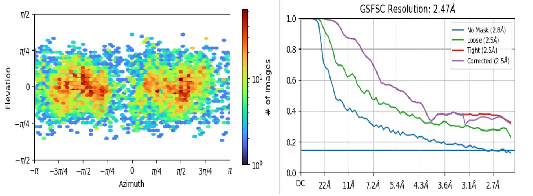
Our data shows a resolution of about 2.5 after completing Homo refinement, but the structure is actually not clear, and we can’t see any fine structures such as amino acids, secondary structures, etc., and we make a mask to estimate the resolution according to the color, and the resolution should be 7-8, so why does Homo refinement display resolution 2.5?
Hi @Heqixin! I agree with @rbs_sci that this looks like duplicate particles. Gold-standard FSC (GSFSC) filtering relies on the assumption that the particles in each half-set are distinct, but if there are duplicate particles it’s possible for the same particle to end up in each half-set (since there are multiple images of that particle).
Thank you very much for your answer. Yes, because my protein is rod-shaped, I use filaments to track the selected particles, and duplicate particles are not removed during two-dimensional classification, so there are a lot of duplicate particles. Do you have any suggestions for my post-resolution characterization if I don’t want to remove duplicate particles too much?Thank you again for you answer!
Yes!because my protein is rod-shaped, I use filaments to track the selected particles, and duplicate particles are not removed during two-dimensional classification, so there are a lot of duplicate particles. Do you have any suggestions for my post-resolution characterization if I don’t want to remove duplicate particles too much?Thank you again for you answer!
You absolutely must remove duplicates. If it is helical, treat it as helical (which opens up a whole new collection of things to worry about) but if not, then blasting reconstructions with duplicate picks will result in pathological FSC curves as you displayed above. If the work made it to manuscript submission, any reviewer with experience would immediately notice and raise concerns.
If you want to use “standard” SPA, you can start by removing particles fairly close (say, 30Å, depending on your particle size - if it’s small, 30Å may be sufficient, if it’s large, then you may need to increase significantly) and seeing how the FSC curve improves. You want to see an FSC curve drop to zero, and stay there.
If uncertain, or very new, I would recommend going through the CryoSPARC tutorial datasets a few times (later runs you can tweak settings to see what happens) and also trying other tutorial datasets (e.g.: RELION tutorial data also processes nicely in CryoSPARC, although the cisTEM dataset is a little too small) or downloading 50-100 micrographs from a few different (“easy”) datasets from EMPIAR like beta-galactosidase (many options, e.g. 10950) or apoferritin (e.g. 10216) and experimenting.
If you go the helical route, there are a lot of potential pitfalls for the unwary or inexperienced, so go through the CryoSPARC helical tutorial in detail (also try an EMPIAR dataset (e.g.: 10306))…