Hi
I’m relatively new to the Single Particle Analysis pipeline, but I’m currently working on processing an icosahedral virus structure with a peptide attached to its surface.
I did a homogenous refinement with ‘I’ symmetry applied and then supplied this refined structure as an input to another homogenous refinement with symmetry relaxation via maximization to figure out the symmetry-breaking feature, i.e., the peptide.
*The parameters that I used for homo refinement with 'I" symmetry applied are:
symmetry: I
minimize over per-particle scale: true
Fit spherical aberration:true
Fit Tetrafoil: true
optimize per particle defocus: true
FSC: J201(2.47A)
*The parameters that I used for homo refinement with symmetry relaxation are:
symmetry:I
Initial low-pass resolution: 20
Symmetry relaxation method: maximization
FSC:J210 (3.79A)
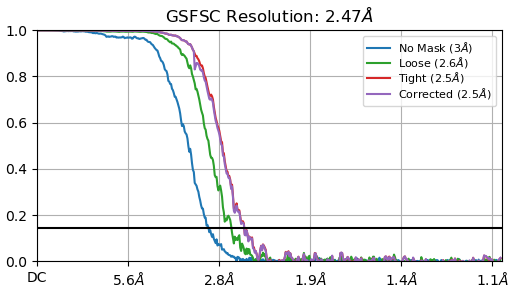
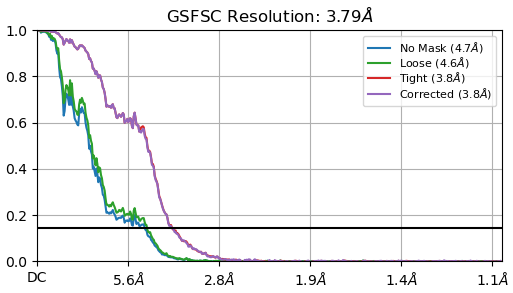
a. Are these parameters accurate? Additionally, I find the FSC curves difficult to interpret; they look so different than the standard curves that I observed in the literature.
b. At the moment, I can’t discern the density of the attached peptide. I intend to perform local refinement to see if I can figure out the density. Do you have any alternative suggestions for this approach?
Thank you
K.S
Hi,
I would recommend using a much less aggressive initial low pass filter for your symmetry-relaxed refinement - based on your first FSC, I would say an initial lowpass of 5Å should be fine. I would also add 5-10 additional final passes (as the convergence criteria are FSC based, they do not yet account for convergence of the sym relaxation procedure).
As an alternative strategy, you might try symmetry expansion followed by classification without alignments with a focus mask around a single subunit, and a solvent mask covering the entire capsid.
For your first refinement, for a large particle like a viral capsid it might also be worth attempting refinement of anisotropic magnification.
Cheers
Oli
Edit: Thinking about this a little further - if what you really have is a peptide stochastically binding/unbinding to individual subunits, then I suspect symmetry expansion followed by classification might be the way to go. Because of the high symmetry, the size of your stack will increase 60-fold, so it might be worth either downsampling and cropping the original particles as much as you can get away with for the purposes of speed, or re-extracting subparticles after symmetry expansion in a much smaller box centered on the focus mask (after re-centering using volume alignment tools).
1 Like
Hi @Kit1234567890,
b. At the moment, I can’t discern the density of the attached peptide. I intend to perform local refinement to see if I can figure out the density. Do you have any alternative suggestions for this approach?
In addition to Oli’s good suggestions, I would also suggest the following masking considerations, for symmetry relaxed refinements:
- It may be worth the disablement of dynamic masking, which can either be done by:
- connecting a generous static mask to refinement, that covers both the virus and the (expected location of the) peptide to ensure that the peptide density isn’t masked away by the dynamic masking procedure.
- Alternatively, you may set the resolution to start dynamic masking parameter to a small value, like 1Å, to prevent masking at all.
- Rather than entirely disable dynamic masking, it may also be worth trying more generous Dynamic masking parameters:
- A lower threshold (e.g. 0.15 instead of 0.2)
- A greater near and far distances (e.g. 10 and 25 respectively, instead of 6 and 14)
- A high start resolution (e.g. 5 Å instead of 12 Å)
Additionally, I find the FSC curves difficult to interpret; they look so different than the standard curves that I observed in the literature.
It would be expected that the symmetry enforced curve looks a lot better than the symmetry-relaxed curve due to the 60-fold increase in the number of observations used during reconstruction.
Best,
Michael
2 Likes