My particles are tetramers and each protomer can bind a small protein inhibitor. However, it seems that my sample was heterogenous and based on 2D classes I am sure I have “apo-tetramers” and also tetramers that only have 1 inhibitor bound. I am not sure if I have tetramers with 2 or 3 or 4 inhibitors bound as well.
On top of this, my sample was quite concentrated and the tetramers are really close to each other. Which means, that I either have the inhibitor sticking out of my tetramer in the close proximity or I have the neighbouring tetramer hanging closeby to my tetramer. This creates a problem where I am unable to select between these two groups.
What should I do in 3D to distinguish between tetramer+second tetramer closeby and tetramer+inhibitor?
I have a different dataset with similar partners where I was able to reconstruct the map for tetramer+1inhibitor by adding the focus mask of protomer+1inhibitor. However, there I didn’t have such a concentrated sample so the map for inhibitor was not “overlapped” with the map for the neighbouring tetramer.
Hope I am making sense and someone can help me figure this out?
Thank you,
Andrea
For 3D classification, what is your best refinement job going in ?
Sometimes Ab-initio with multiple classes, then heterogeneous refinement of that can really get this out.
3D classification will take some optimization, but you can filter down do an appropriate resolution (6-12A) ? Depends on the size of the inhibitor. For this you can also use the solvent mask from refinement. Force Hard Classification = on and O-EM learning rate init = 1 can bring out the diverse stuff. Increasing Number of O-EM epochs could help a little.
I did not try Topaz, but I tried template picking.
Best refinement is about 2.8A from 1.3 mil particles (but the map in the area of inhibitor is a mix between the inhibitor and the neighbouring particle). I used C1, because I think I only have 1 bound inhibitor, even though for tetramer C2 works better.
I did 6A (my inhibitor is about 15Da). I did force hard classification. But I decreased the learning rate, which I see was wrong.
Will try again with higher epochs, learning rate 1.
If there are a sufficient number of classes and the learning rate is increased 0.4=>1 that should bring out the most diverse.
Applying symmetry may average out particles with different amounts of inhibitors bound.
I think TOPAZ, if trained correctly can pick better than template. The CryoSparc team also suggests that template and blob picking on denoised micrographs yields different results, which is also true for TOPAZ (topaz has its own denoiser, CS uses its own for blob and template).
15 kDa inhibitor should be easier.
You will be surprized by diversity of 3D classes with learning rate =1. To quickly see if a 3D class is promising, just take the particles for Homogeneous Reconstruction and see how that goes. May need to flip the hand. If you have two inhibitors on in the 3D class with C1, you could try enforcing C2 symmetry.
I would recommend enforcing C2 for refinement, then separating bound/unbound with 3D classification on a symmetry expanded set, with a focus mask around one protomer (or tighter around the inhibitor binding site).
In our experience this has been the most robust way to address such situations - I think you benefit in classification from the more accurate poses that you will get from a C2 refinement.
If this is a compositional case with a 15kDa inhibitor, you might start with a filter res of 10 or 12 Å, maybe 3 or 4 classes, fix learning rate at 1 and tweak from there as needed.
I started the 3D classification. It did 97 iterations by now and it took 27840 seconds. It wants to do a total of 3771 iterations. So I guess in 12 days I will know how did it go
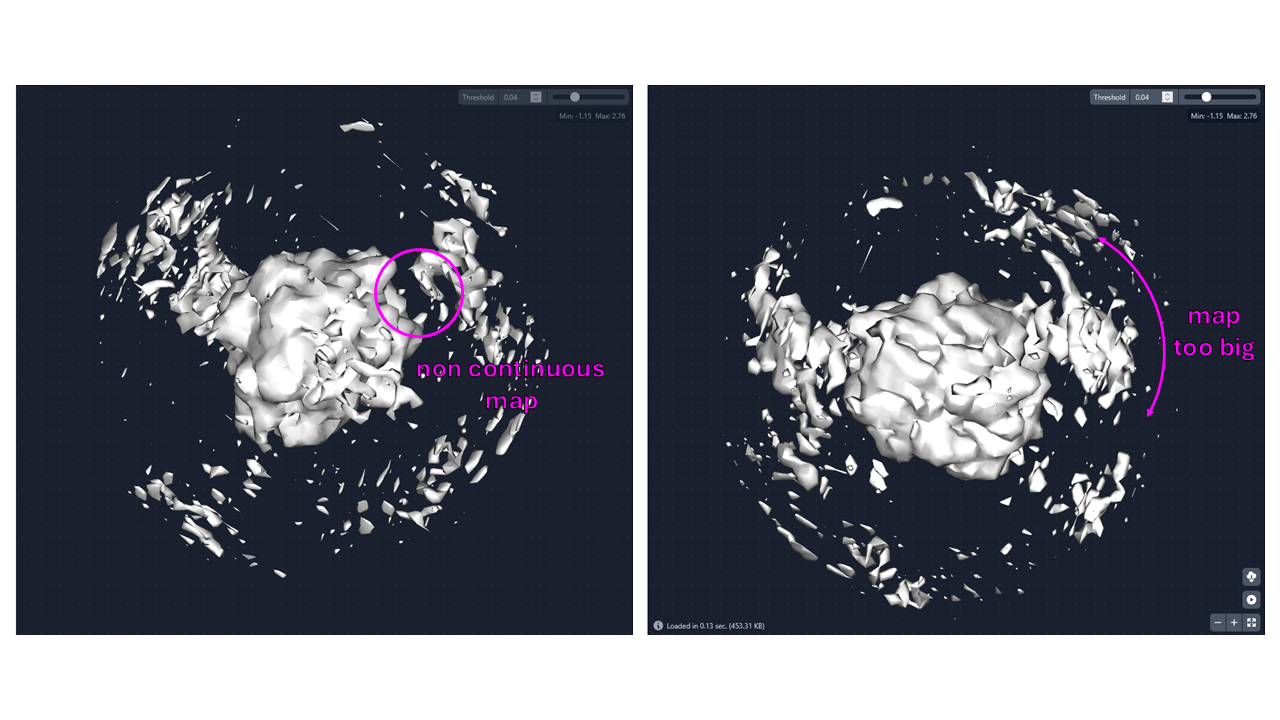
I tried 3D with 200k particles and 90.8% ended in 1 class with 5.2%, 2.5% and 1.5% in the others. The volumes don’t look much different though. There are blobs in all of them kind of where I expect my inhibitor, but the map is not continuous and it is too big/broad. So the 3D can’t distinguish between my inhibitor and the neighbouring particles, just putting all of it superposed in one map…
For the refinement gonig into 3D classification, did you be chance change anything in the Window inner/outer radius? or anything with Dynamic Mask Far (A) ?
You could get around this with extracting at a different box size, some re-centering by center of mass, and later on some masking.
I need a bigger box size so I can capture the inhibitor when/if it is there. But I either get the inhibitor or part of the next particle because the inhibitor and the particle can be in the similar/same spot.
Yes, I used inner/outer radius. I didn’t use dynamic mask far.
good that you have tried the different box sizes and experimented with inner/outer window for refinement.
Is it possible the inhibitor oligmerizes the protein ? Do you have any biophysics to support the distribution in solution (SEC/SEC-MALS, DLS, SA(X/N)S, AUC, Mass Photometry) ? It could just be as your first post…