Hi,
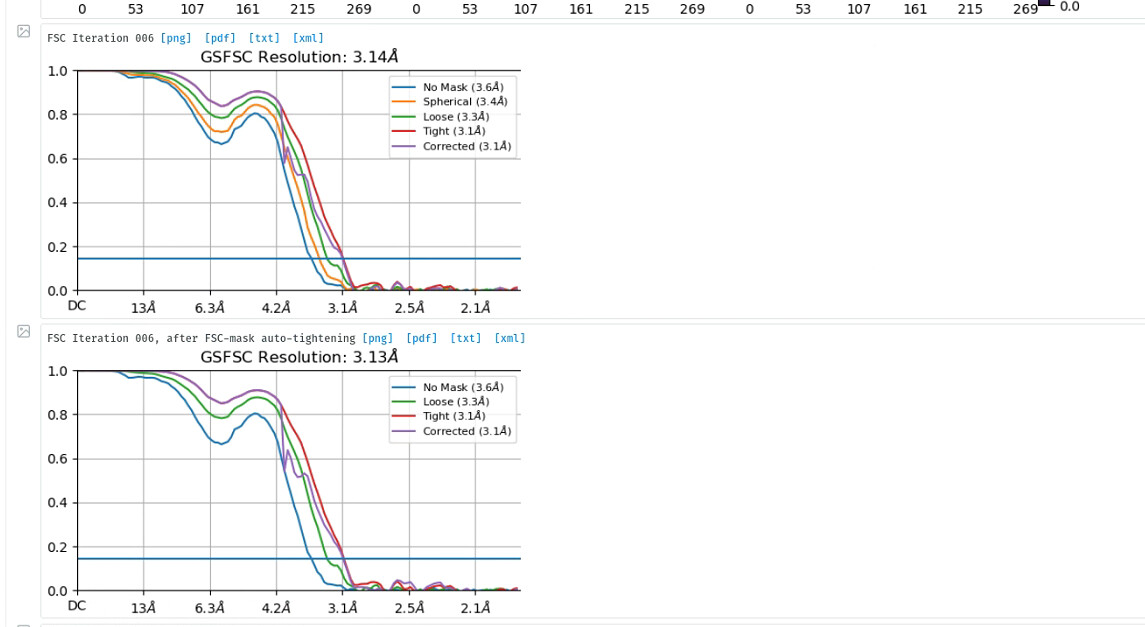
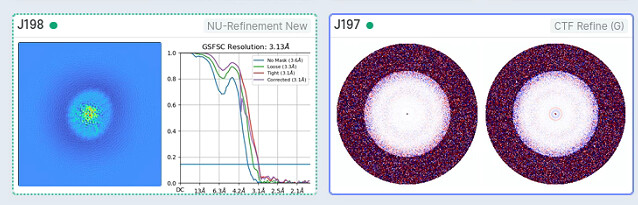
After NU refinement with 240k particles, dynamic mask, 3.14A, I want to do local refinement to improve resolution. But I find the resolution limited to 3A even I masked out micelle. The local resolution estimation result shows that the main body of protein is about 2.9A, I don’t know how to improve the resolution further and what can I do next. Can anyone help me? Any advice is appreciated.
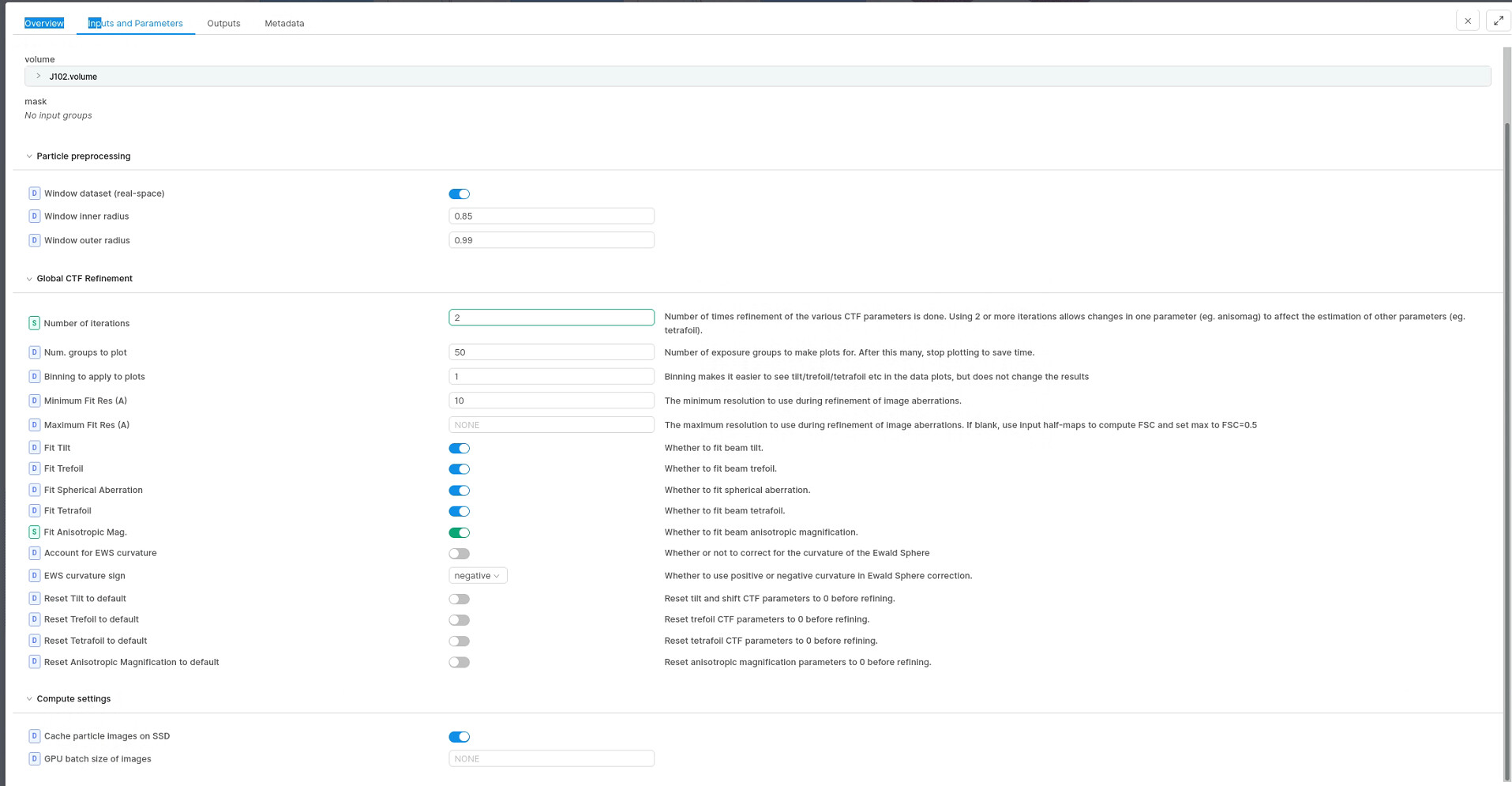
What does your FSC look like? Have you tried Global CTF refinement (refining beam tilt & trefoil)? It may be that something else is limiting your resolution, or it may be just that your sample is intrinsically somewhat dynamic, or has unresolved compositional or conformational heterogeneity.
It’s really tough to say but likely those red regions of lower resolution are flexible, and so you could consider masking those out while you refine the rest of the structure, but it’s questionable what that would do that the NU refinement didn’t.
Is the reference the same particle? What part of the particle were they doing the local refinement on? Could you link the paper?
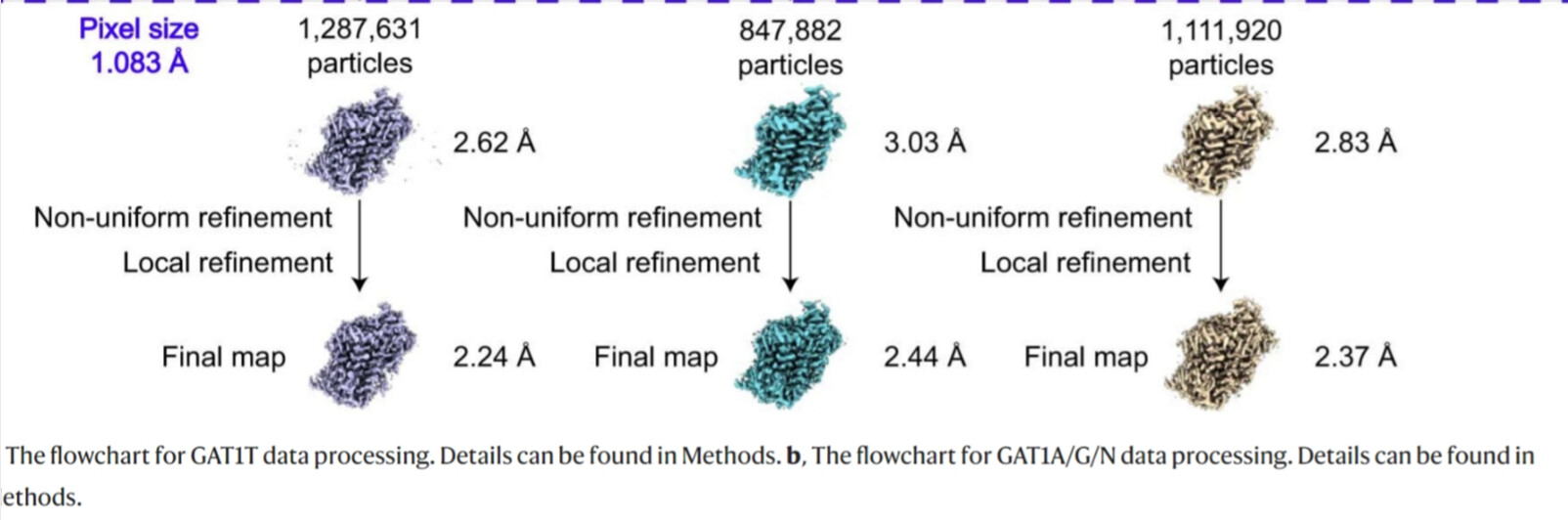
I am also intrigued by its remarkable improvement in resolution. Here are the original figures: https://www.nature.com/articles/s41594-023-00983-z/figures/8
By the way, it appears that the structures from the paper mentioned above lack any cytoplasmic components.
I work on similarly sized membrane proteins, and I found that CTF refinement doesn’t really do anything. I tried various settings, at various steps but it never improved my resolution. Tho the highest res. I obtained was 3.2A.
I am also surprised by the local refinement, it usually helps me only by 0.2A, although I am also not sure if that isn’t just an inflated FSC. I barely see any improvement to the density. Something that does help me a lot is: minimization over particle scales, and testing if multiple full iterations are needed or not (you can observe it in the event log), and at this stage, bayesian polishing might improve your resolution.
Thanks a lot. My particles are polished before NU refinement. Do you do local refinement just masking out micelle? What do you mean about minimization over particle scales and multiple full iterations? Are those local refinement parameters?
Yes, if I do local refinement, I create a mask that excludes the micelle.
Both NU and Local refinement have two sliders for: Reset input per-particle scale (I turn this off) and Minimize over per-particle scale (I turn this on). There is also a setting called Number of extra final passes, which I try to figure out if I need to increase it. Don’t go crazy with it, otherwise you might end up over-fitting, but check if you need to increase it a little bit further. On the event log, you can observe if there is a trend and your structure might benefit from yet an other iteration or not.
And for local refinement, I use gaussian priors (on) and keep the two standard deviations numerically low
Global CTF refinement will help, sometimes dramatically, if there are aberrations to correct - if not, it won’t do anything. Sounds like you have a happy & well aligned scope!
Local CTF (defocus) refinement will help if you have a larger particle in thick ice - for small particles in thin ice it is unlikely to make much difference except at very high resolution.
The effect of local (masked) refinement will vary substantially depending on the case - i.e. how much variability is present outside the mask. In some cases I have seen dramatic improvements, in other cases minimal. In certain cases careful exploration of mask parameters (softness, size) and search parameters is needed to achieve optimal results.
Hi, Oli. Thanks for your suggestion. I wonder which search parameters you would often change to achieve better results? I’ve tried changing following parameters but it doesn’t work dramatically.