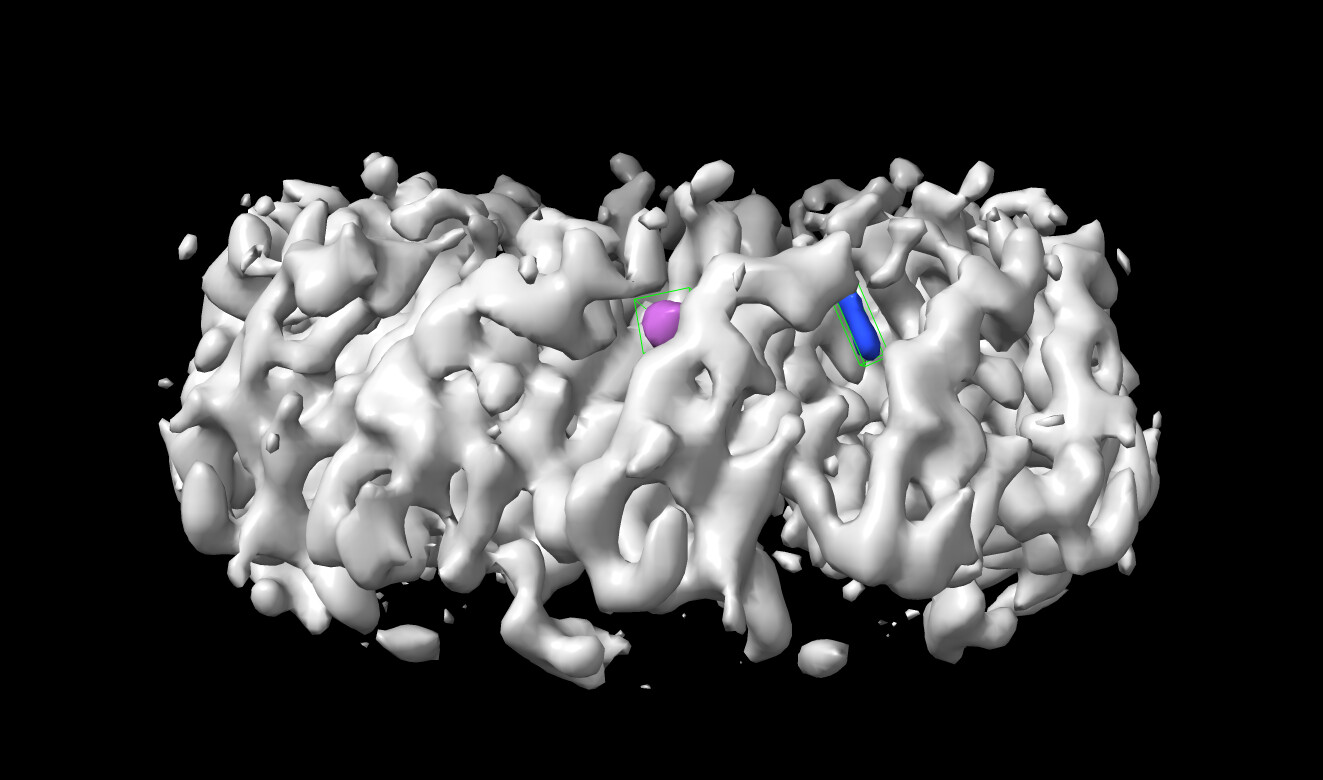
I am working on a hexameric, ring-shaped protein with C6 symmetry. Each protomer of the protein contains a linked dimer. The protein has totally 12 binding sites but every other site is mutated to be unable to bind to the ligand so I am expecting to see the density for the ligand at every other site in the final refined map. After 3D refinement, the final map resolution is ~4 Å. However, I found some unexpected density at the mutated binding site (unable to bind to the ligand). In the attached image, the density of the ligand is colored blue and the unexpected density is colored purple. We are concerned that this unexpected density might be caused by image misalignment. I am wondering if anyone could provide some suggestions on how to identify if this is a misalignment issue and how should I correct it.
If all else besides ligand site is the same between the two parts of the dimer, then it’s highly possible that alignment is driven by the huge amount of identical density and not the tiny ligand. In either case; either make a distinguishable difference in one of the two, or collapse the 6 dimers with symmetry expansion and see that one site is liganded and the other does(not) have density. For this you may need to collapse all 12 if they’re the same and classify with mask for presence of ligand.
Also, apply symmetry to get a higher resolution map, use as reference, 3D classification beta, etc. 4A is not ideal to interpret presence/absence of this small density
In addition you could try C3 symmetry, as I understand it - It is not true C6 but C3 with dimers that are different at the binding site. Or expand the particles with C3 and then try local refinement on one dimer with the search range a little bit more than the distance between the two differently mutated binding sites, that might sort out any image misalignment.
Another thing you could try is to do supervised 3D classification where you give in the dimer with and without the extra densities and see if it can sort different classes. Altough I suspect this density might be too small to do that on the full particles, but if you first do local refinement, and then do the 3D classification you might get a pretty good idea.