I wanted to share my current strategy for high-throughput particle picking with crYOLO and cryosparc-tools, using SLURM on an HPC cluster, just in case anyone else is ever trying to do this, or perhaps others have tips to share as well. You can also just set slurm_array = False and use this as a convenient Python crYOLO/CryoSPARC script, that’s easily editable since all the variable are at the top.
I’ve adapted this script from what is posted here:
2. Pick particles with crYOLO — cryosparc-tools
Most of this script is not my own.
To get started, see the example cryolo.py at the bottom of this post, as well as the cryolo.sbatch for the SLURM cluster submission script. You’ll need a crYOLO module with cryosparc-tools installed on your cluster.
-
In
cryolo.pyfill out the CryoSPARC Master settings. -
Make a job that contains your 5-10 training micrographs and particle locations. I normally use a “Curate Exposures” job for this, and reject the micrographs and particles I want to use. (There’s no reason for this except that it’s convenient). In this example, that’s Job 291, and the important outputs are
exposures_rejected' andparticles_rejected`.
In the Job Settings portion of cryolo.py, set the project and workspace. Set training_micrographs to the job that has the 5-10 training micrographs, and set training_micrographs_output to the name of the output group that has them (in this case, rejected_exposures). Do the same for the particle groups.
# Job Settings - Replace n with number
job_title = "crYOLO Picks" # Title for your job
cs_project = "P38" # Project number
cs_workspace = "W6" # Workspace number to create jobs in
training_micrographs = "J291" # Job number with output that is to be used as input for training
training_micrographs_output = "exposures_rejected" # Which job output group to use (can get from job metadata if needed)
training_particles = "J291" # Which job contains the particles to use for training
training_particles_output = "particles_rejected" # Which output from that job to use for training particles
- crYOLO picking tends to fail for me for jobs > 1000 micrographs or so. Because I’m using BIS data collection, I just split up my exposures into exposure groups using the Exposure Groups Utilities tool. Since this was a 5x5 data collection, I have outputs
exposure_group_0throughexposure_group_24
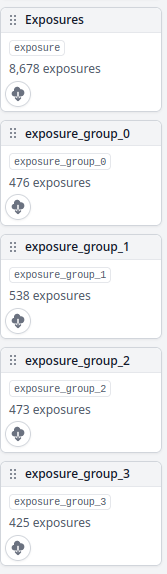
In Job Settings in cryolo.py, set picking_micrographs to the job number containing the micrographs you’d like to pick. If you are using exposure groups, set picking_micrographs_output to exposure_group and leave the _0 or other number suffix off. SLURM will add that automatically for each exposure group. If you are only doing one output group, just set it to the name of the output group.
picking_micrographs = "J290" # Which job contains the micrographs to pick
picking_micrographs_output = "exposure_group" # Which output from that job to use
-
If you have multiple exposure groups, SLURM can automatically increment the number at the end, and automatically submit a job for each exposure group. This is very handy becaue then we don’t have to submit 25+ different jobs. Simply set
slurm_array = Trueto enable this. -
Setup your SBATCH script. The important thing here is to add an SBATCH parameter for arrays. Here is mine:
#SBATCH --array=0-24. You’ll want to set this to be the numbering of the exposure groups. If you used the Exposure Group Utilities tool, CryoSPARC will output them starting at 0, and ending with the number of exposure groups-1, so for my 5x5, that’s 0 through 24.
For the submission line, simply call the Python script, but with the variable ${SLURM_ARRAY_TASK_ID} as the first argument:
python cryolo_sample_5.py ${SLURM_ARRAY_TASK_ID}
This will generate as many jobs as you set your array to (so as many jobs as exposures groups), and for each job, it will automatically increment the exposure group to pick by 1.
- Launch the SBATCH script. You will see SLURM queue up your jobs using as many GPUs as it has access to:
As one job finishes and a GPU frees up, it’ll launch another job automatically.
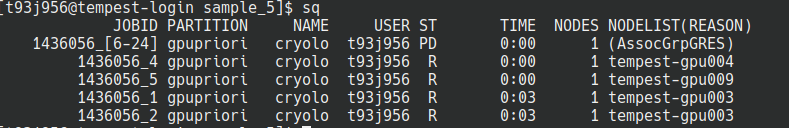
Check CryoSPARC to see that it’s working:
Python script (cryolo_sample_5.py):
import sys
from cryosparc.tools import CryoSPARC
from io import StringIO
import numpy as np
from numpy.core import records
from cryosparc import star
from cryosparc.dataset import Dataset
##################################################################################################################################################
# VARIABLES VARIABLES VARIABLES #
##################################################################################################################################################
# CryoSPARC Master Settings
cs_host = "mycrosparc.mycampus.edu" # URL of CryoSPARC Master instance
cs_port = 39000 # Port of CryoSPARC Master instance
cs_email = "myemail@mycampus.edu" # Email address of your CryoSPARC account
cs_pass = "mypassword" # Password for your CryoSPARC account
cs_license = "mylicensenumber" # License key for your CryoSPARC instance
# Job Settings - Replace n with number
job_title = "crYOLO Picks" # Title for your job
cs_project = "Pn" # Project number
cs_workspace = "Wn" # Workspace number to create jobs in
training_micrographs = "Jn" # Job number with output that is to be used as input for training
training_micrographs_output = "exposures_rejected" # Which job output group to use (can get from job metadata if needed)
training_particles = "Jn" # Which job contains the particles to use for training
training_particles_output = "particles_rejected" # Which output from that job to use for training particles
picking_micrographs = "Jn" # Which job contains the micrographs to pick
picking_micrographs_output = "exposure_group" # Which output from that job to use
# crYOLO Settings
threshold = "0.3" # Threshold for crYOLO picker - defaults to 0.3. Lower to get more picks, raise to get fewer picks.
gpu_number = 0 # Enter "0" to use 1st GPU, "1" to use 2nd GPU, or "0 1" to use both GPUs
#SLURM Settings
slurm_array = True #Set to "True" if using a SLURM array to submit multiple jobs.
#################################################################################################################################################
if slurm_array == True:
print("SLURM array set to true. Submission will be: ")
picking_micrographs_output = picking_micrographs_output + "_" + str(sys.argv[1])
print(picking_micrographs_output)
else:
print("Standard submission - no SLURM")
#Setup CryoSPARC Connection
cs = CryoSPARC(host=cs_host, base_port=cs_port, email=cs_email, password=cs_pass, license=cs_license)
assert cs.test_connection()
#Setup Job
project = cs.find_project(cs_project)
job = project.create_external_job(cs_workspace, title=job_title)
job.connect("train_micrographs", training_micrographs, training_micrographs_output, slots=["micrograph_blob"]) #Micrographs to use as input to training network
job.connect("train_particles", training_particles, training_particles_output, slots=["location"]) #Particles to use as input to training network
job.connect("all_micrographs", picking_micrographs, picking_micrographs_output, slots=["micrograph_blob"]) #Micrographs to pick from
job.add_output("particle", "predicted_particles", slots=["location", "pick_stats"]) #Initialize output
'predicted_particles'
job.start() #Initializes an external job
#Create necessary directory structure
job.mkdir("full_data")
job.mkdir("train_image")
job.mkdir("train_annot")
all_micrographs = job.load_input("all_micrographs", ["micrograph_blob"])
train_micrographs = job.load_input("train_micrographs", ["micrograph_blob"])
for mic in all_micrographs.rows():
source = mic["micrograph_blob/path"]
target = job.uid + "/full_data/" + source.split("/")[-1]
project.symlink(source, target)
for mic in train_micrographs.rows():
source = mic["micrograph_blob/path"]
target = job.uid + "/train_image/" + source.split("/")[-1]
project.symlink(source, target)
job.mkdir("train_annot/STAR")
train_particles = job.load_input("train_particles", ["location"])
for micrograph_path, particles in train_particles.split_by("location/micrograph_path").items():
micrograph_name = micrograph_path.split("/")[-1]
star_file_name = micrograph_name.rsplit(".", 1)[0] + ".star"
mic_w = particles["location/micrograph_shape"][:, 1]
mic_h = particles["location/micrograph_shape"][:, 0]
center_x = particles["location/center_x_frac"]
center_y = particles["location/center_y_frac"]
location_x = center_x * mic_w
location_y = center_y * mic_h
outfile = StringIO()
star.write(
outfile,
records.fromarrays([location_x, location_y], names=["rlnCoordinateX", "rlnCoordinateY"]),
)
outfile.seek(0)
job.upload("train_annot/STAR/" + star_file_name, outfile)
job.subprocess(
(
"cryolo_gui.py config config_cryolo.json 130 "
"--train_image_folder train_image "
"--train_annot_folder train_annot"
).split(" "),
cwd=job.dir(),
)
train_string = "cryolo_train.py -c config_cryolo.json -w 5 -g %s -e 15" % (gpu_number)
job.subprocess(
# "cryolo_train.py -c config_cryolo.json -w 5 -g 0 -e 15".split(" "),
train_string.split(" "),
cwd=job.dir(),
mute=True,
checkpoint=True,
checkpoint_line_pattern=r"Epoch \d+/\d+", # e.g., "Epoch 42/200"
)
job.mkdir("boxfiles")
predict_string = "cryolo_predict.py -c config_cryolo.json -w cryolo_model.h5 -i full_data -g %s -o boxfiles -t %s" % (gpu_number, threshold)
job.subprocess(
# "cryolo_predict.py -c config_cryolo.json -w cryolo_model.h5 -i full_data -g 0 -o boxfiles -t %s".split(" ") % threshold,
predict_string.split(" "),
cwd=job.dir(),
mute=True,
checkpoint=True,
)
output_star_folder = "STAR"
all_predicted = []
for mic in all_micrographs.rows():
micrograph_path = mic["micrograph_blob/path"]
micrograph_name = micrograph_path.split("/")[-1]
height, width = mic["micrograph_blob/shape"]
starfile_name = micrograph_name.rsplit(".", 1)[0] + ".star"
starfile_path = "boxfiles/STAR/" + starfile_name
locations = star.read(job.dir() / starfile_path)[""]
center_x = locations["rlnCoordinateX"] / width
center_y = locations["rlnCoordinateY"] / height
predicted = job.alloc_output("predicted_particles", len(locations))
predicted["location/micrograph_uid"] = mic["uid"]
predicted["location/micrograph_path"] = mic["micrograph_blob/path"]
predicted["location/micrograph_shape"] = mic["micrograph_blob/shape"]
predicted["location/center_x_frac"] = center_x
predicted["location/center_y_frac"] = center_y
predicted["pick_stats/ncc_score"] = 0.5
all_predicted.append(predicted)
job.save_output("predicted_particles", Dataset.append(*all_predicted))
job.stop()
The SBATCH I used - yours will vary depending on your system configuration:
#!/bin/bash
#SBATCH --partition gpupriority
#SBATCH --account priority-cryoem
#SBATCH --nodes=1
#SBATCH --ntasks-per-node=1
#SBATCH --cpus-per-task 16
#SBATCH --gpus-per-task=1
#SBATCH --mem 120000
#SBATCH --time 72:00:00
#SBATCH --job-name cryolo
#SBATCH --array=0-24
#SBATCH --output cryolo-%j.out
#SBATCH --error cryolo-%j.err
module load cryoem/cryolo/1.9.6
python cryolo_sample_5.py ${SLURM_ARRAY_TASK_ID}
Note: one downside is that it’ll re-train the model every time. Since we’re using so few micrographs, though, this goes pretty quick. There’s probably a way to check the root folder to see if a model exists, and just use that, but I haven’t bothered doing it yet.