Hello everyone,
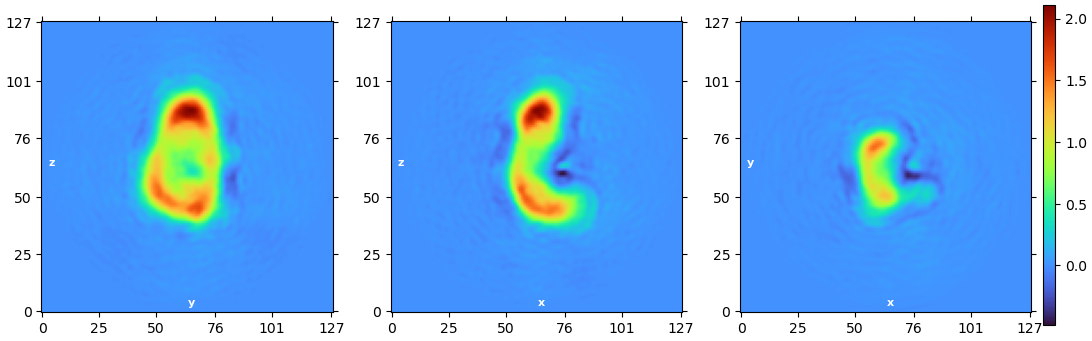
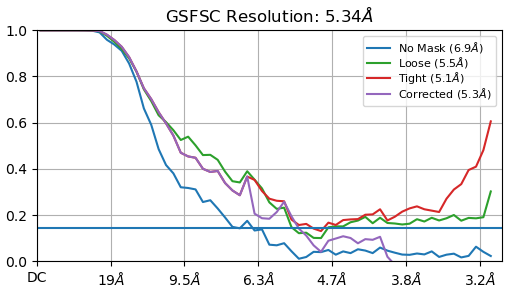
I am having some issues with processing and was wondering if I could get some tips. I am looking at secondary RNA structures and have carried out several heterogeneous refinements before this step. The GSFSC is reporting a 5.34 Å after masking, however when I open it on chimera x it does not look like that.
Has someone had this issue before? Or has any tips?
Thanks
Francesca
None of the 2D classes look very convincing. I would consider reclassifying and asking CryoSPARC to make maybe 100-200 classes. I’d also consider increasing the resolution of the classification to see if you can further separate them.
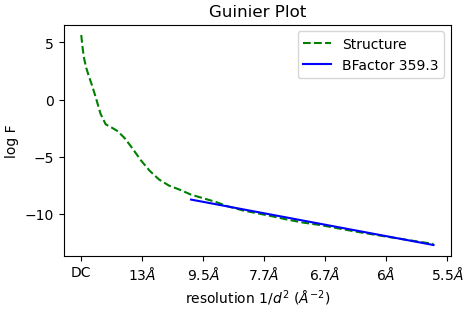
The high B-factor and the FSC not going to zero is strange. Ultimately, it looks like you have a vast amount of conformational variability. Out of curiosity, is this negative stain data?
2 Likes
As @ccgauvin94 says, try a lot more classes (I’d try even more than he suggests - perhaps 300) and perhaps change the “Initial classification uncertainty factor” to something like 5.
2 Likes
The data is from cryo-em data. I’ll try doing a 2D classification with more classes and then report back
These results are typical for RNA-only particles in cryo. Consider that you are imaging a highly flexible object, which is very (very) likely to populate many states in a fast exchange regime. You need to find subsets of all those states that average reasonably well. Have you run native gels with the RNA? Have you screened buffer conditions to obtain the sharpest native gel bands (as recommended by DRRAFTER paper)?
I suggest a couple of things:
- Make sure particle picking is as good as possible, try the EMAN2 neural net picker (very different approach from “deep learning” pickers - note all of the Das lab structures use this picker)
- Try multiple rounds of 2D, always set online EM iterations to a high number, 60-100, use large batch size per class (e.g. 600, with binned particles of course), limit the alignment to 12 Å, try selecting only the large or only the compact particles across a 2-4 rounds of 2D (note many rounds of 2D in the Das lab papers)
- Try ab initio jobs with different numbers of classes, using particles before and after all that 2D
- Good initial references are extremely important, make sure you have stable results from ab initio, limit the resolution of your 3D jobs, only when you are sure you have correct structures you can re-pick with templates (to ensure all views captured)
4 Likes
Hi,
Yes I have run both a native and denaturing and optimised the buffers I am using for folding. I have just been struggling when it comes to 2D as I can’t really see any groves as shown in some papers. Thank you for your suggestions I will give them a try. I have tried cryolo and blob picker for the picking and I reckon cryolo does a better job. I have not tried EMAN yet, but will give it a try. Out of curiosity does the importing of the picking from EMAN need to be converted before cryosparc processing?
Thank you for your help
I can’t remember if I imported them to Relion first or to cryoSPARC, EMAN2 makes .box files.
I think it’s not always possible to get grooves in 2D with these, but hopefully you can see them in ab initio! (And you can try all of Oli’s tips for ab initio, if you search around the forums).
1 Like
Ah right! Thank you, I’ll have a look around for Oli’s post.