Hi everyone,
I’ve got a soluble protein complex (45kDa+55kDa) for cryo-EM study.
Movies: 0.58A, 300V, 40e/A2; Particle Extraction: 256pix->128pix
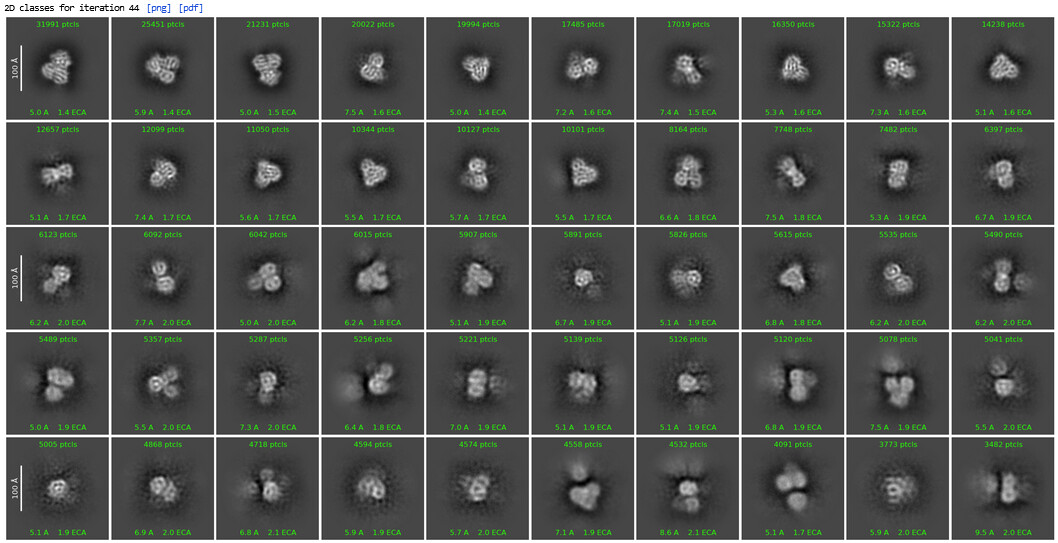
1st 2D Classification
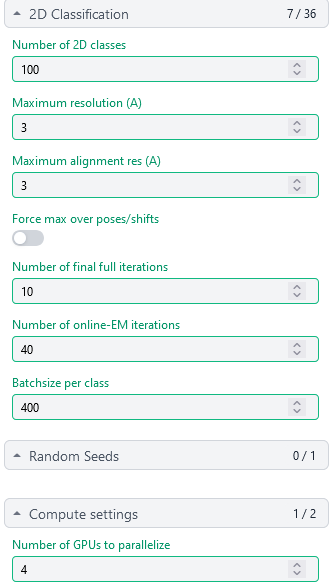
1st Select 2D:get rid of most dummy particles (7,695,260 down to 1,123,196)
2nd 2D Classification, same settings

Could see secondary structures such as a-helices.
2nd Select 2D: (1,123,196 down to 350,114)

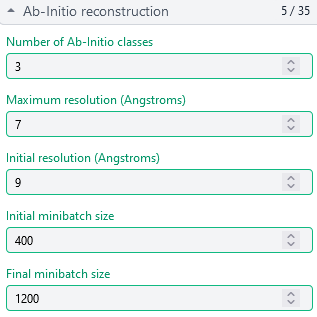
Ab-Initio (3 classes; Particles: 2nd select 2D, 350,114)
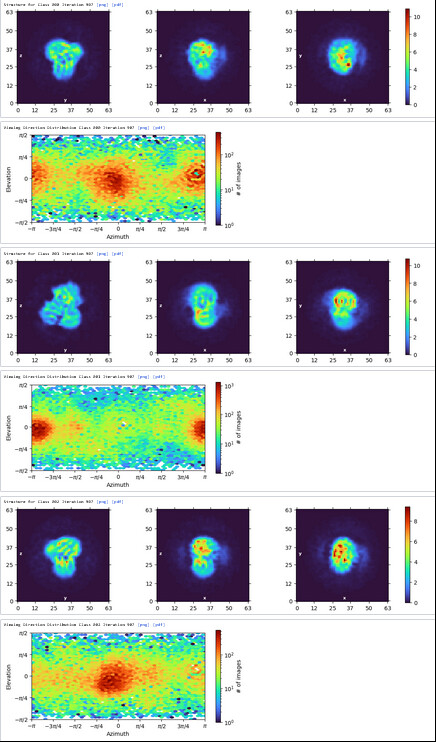
1st Hetero Refinement (Volume: classes0&1 from Ab-Initio job; Particles: 1st select 2D, 1,123,196; Force hard classification)
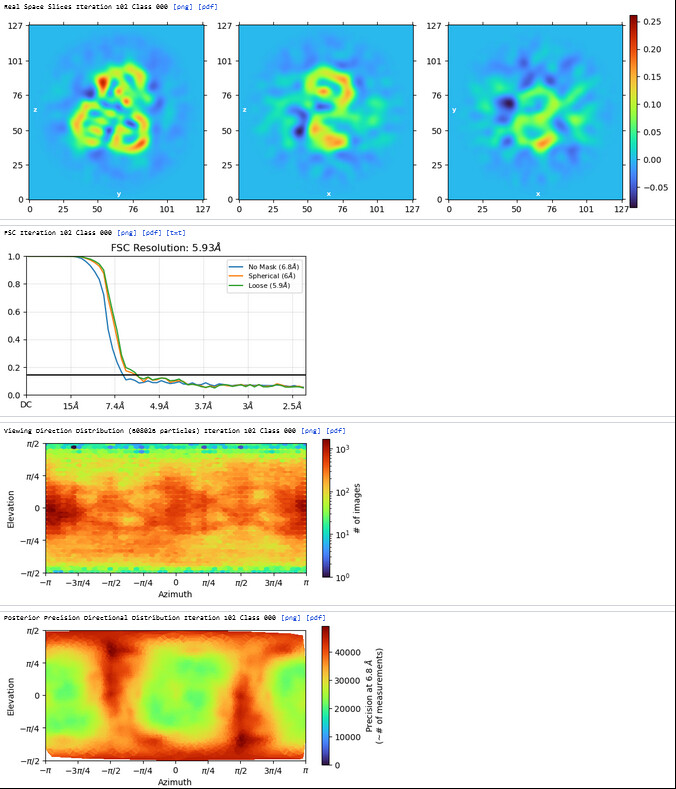
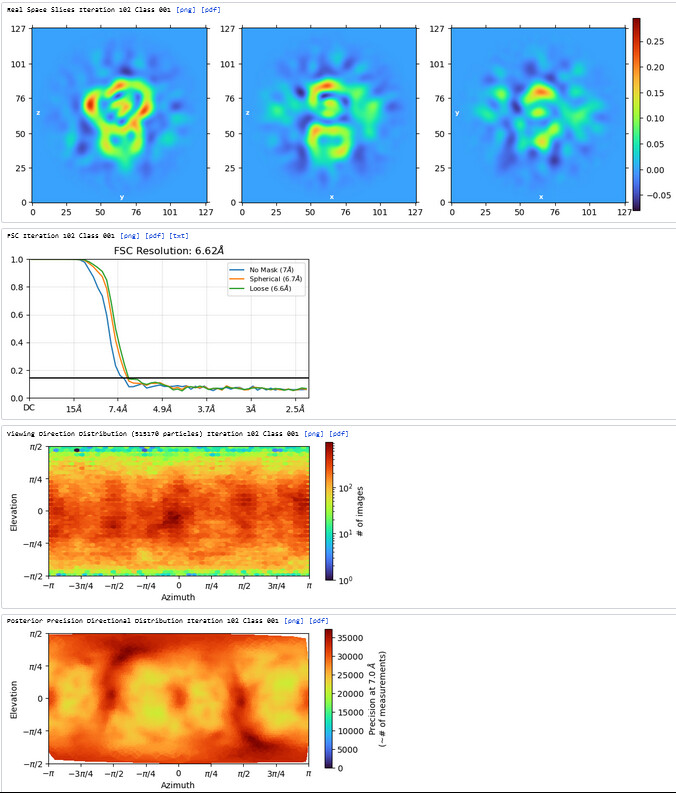
2nd Hetero Refinement (Volume: classes0&1 from Ab-Initio job; Particles: class0 from 1st Hetero Refinement, 608,026; Force hard classification)
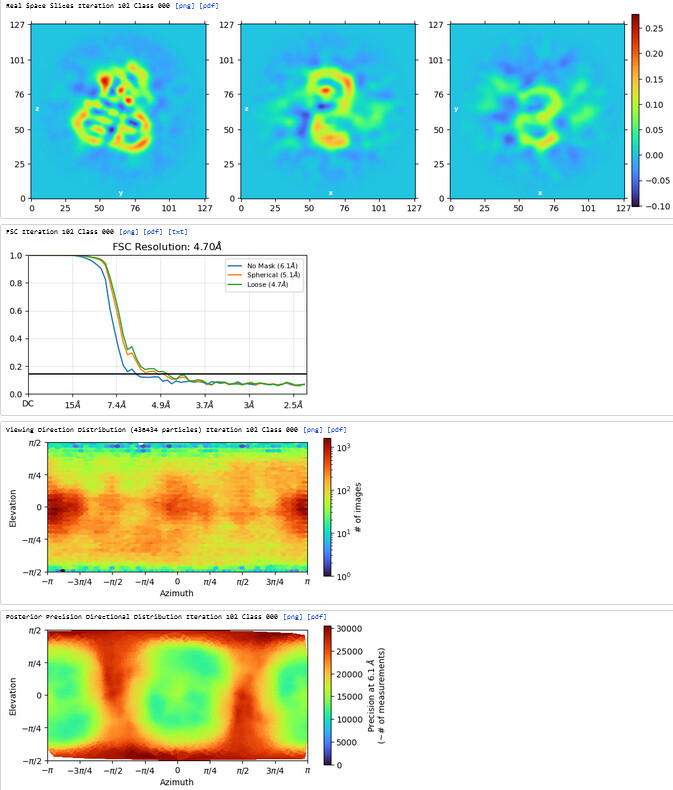
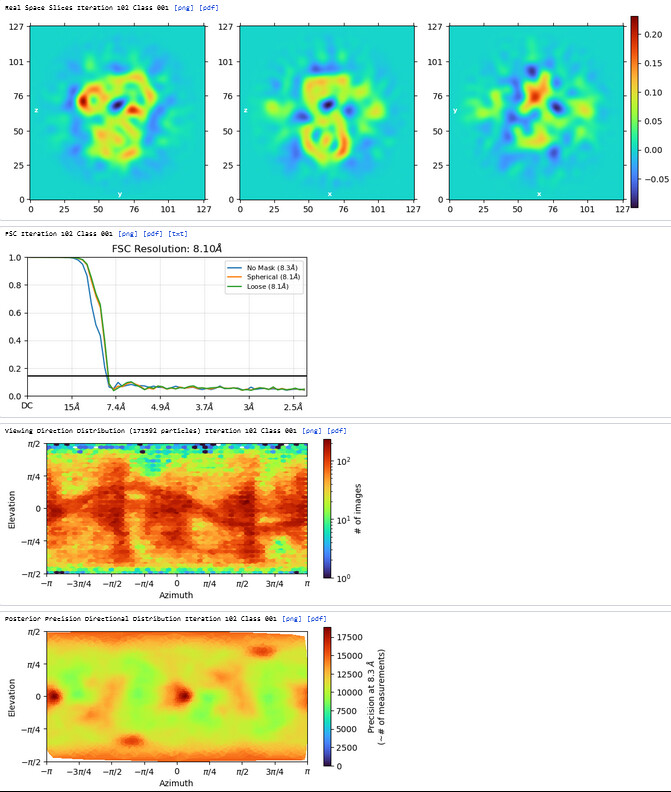
3rd Hetero Refinement (Volume X2: class0 from 2nd Hetero Refinement; Particles: class0 from 2nd Hetero Refinement, 436,434)
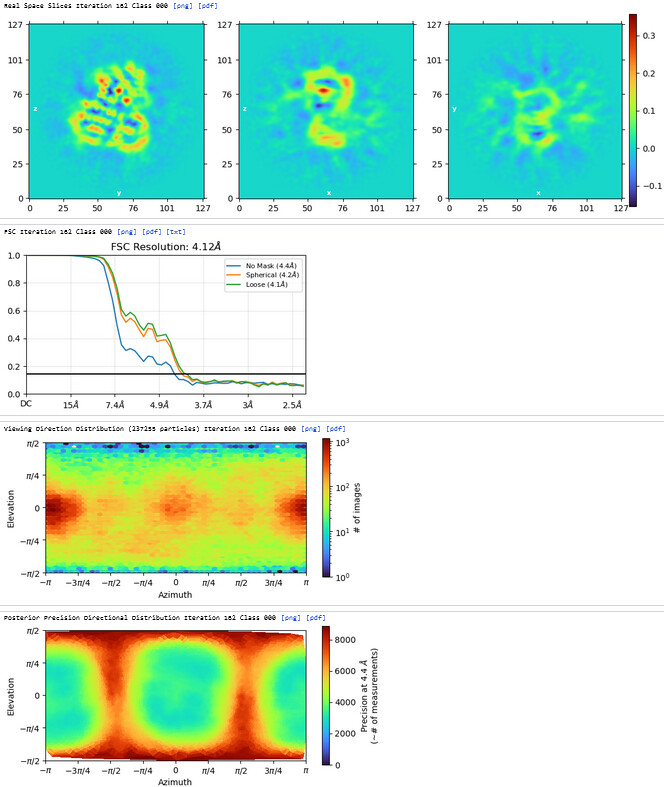
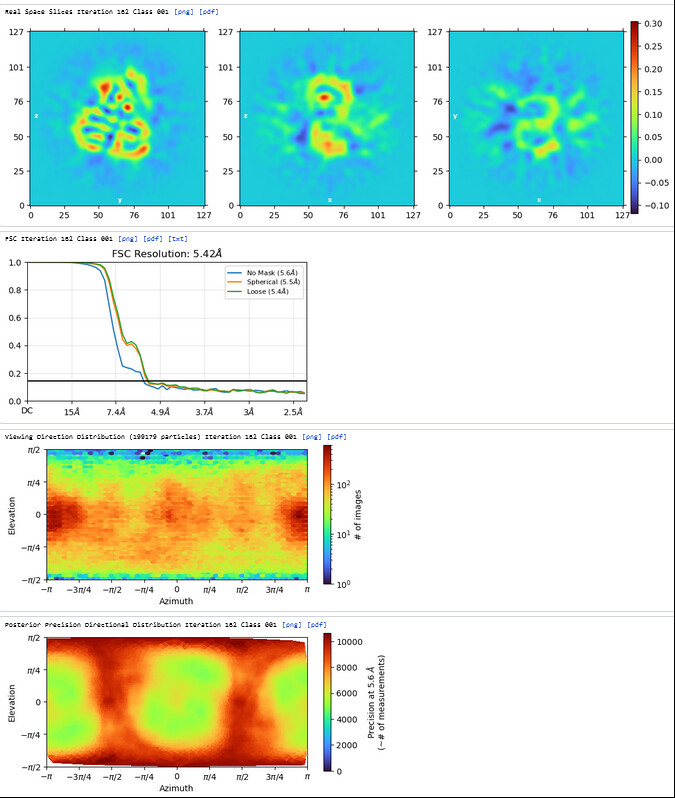
The resolution of class0 could get to 4.1A.
Homo Refine (Volume&Particles: classe0 from 3rd Hetero Refinement, 237,255; Minimize over per-particle scale)
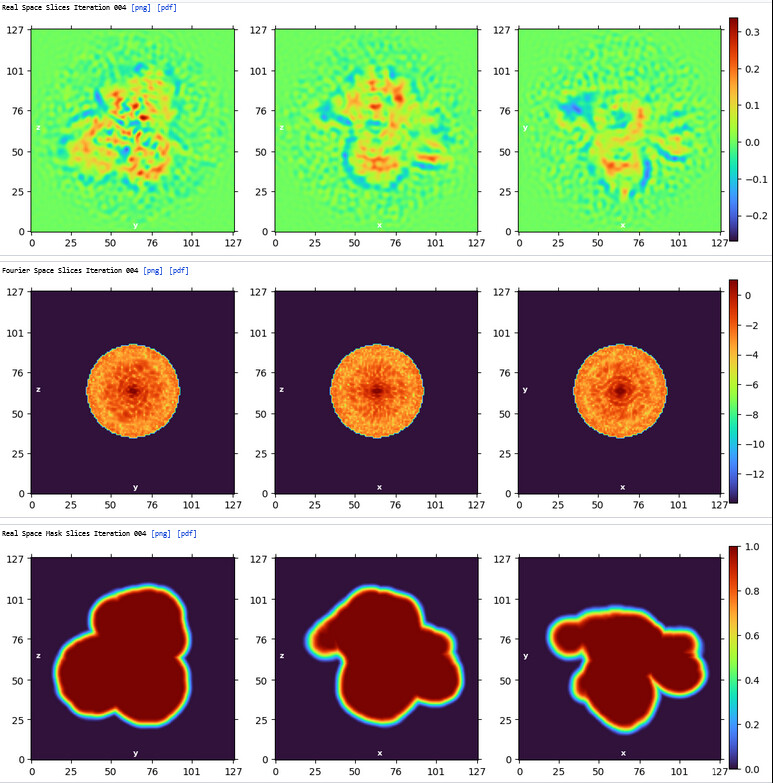
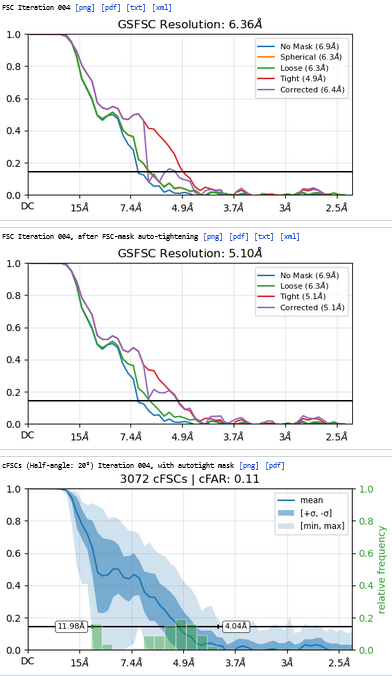
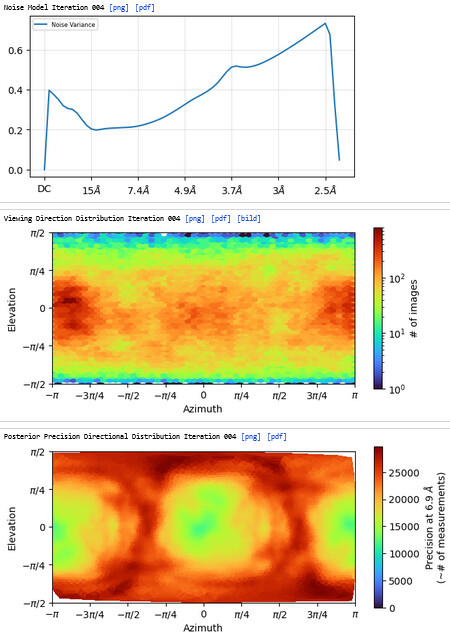
The resolution got worse, even worse than 3rd Hetero Refinement.
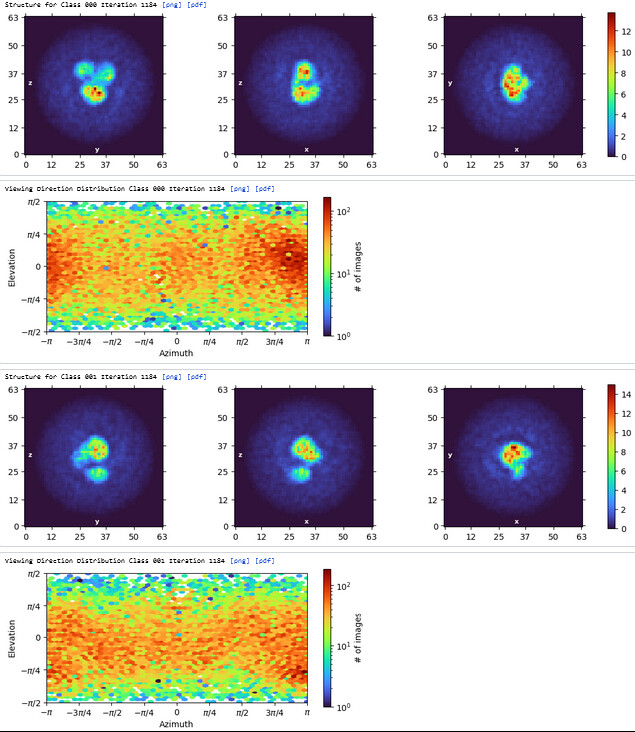
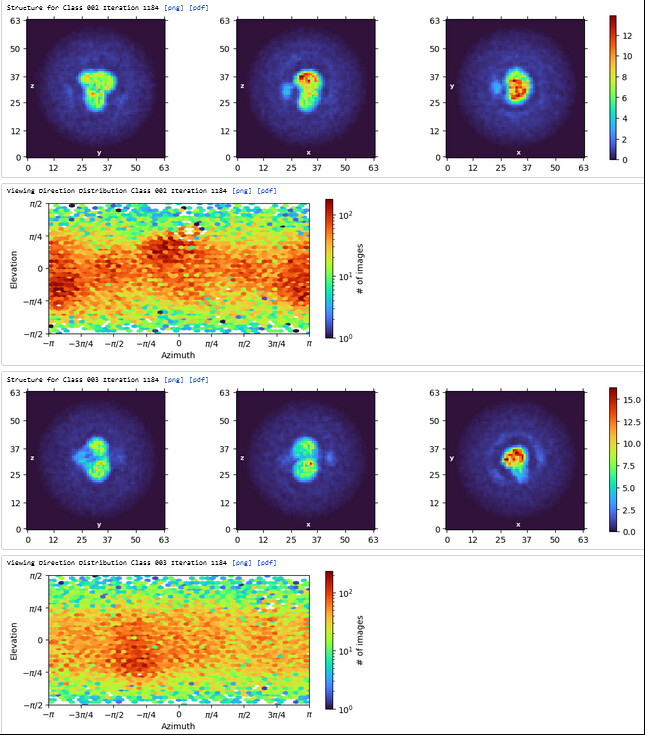
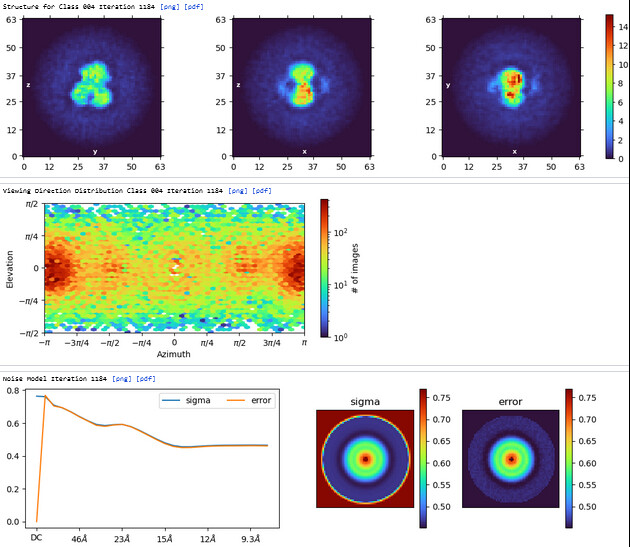
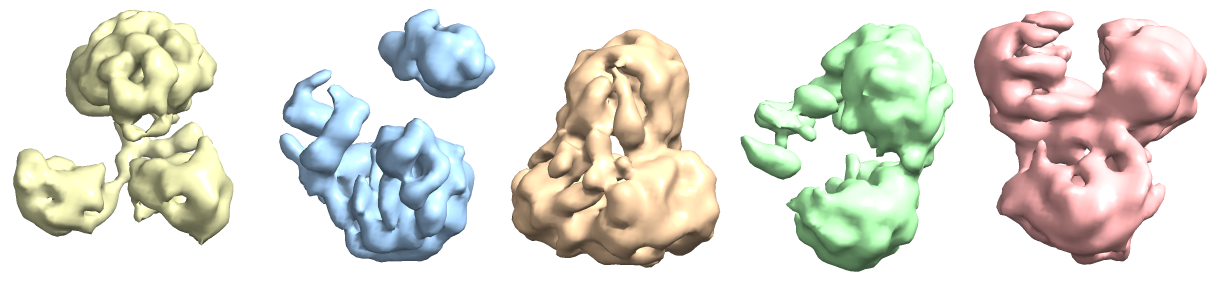
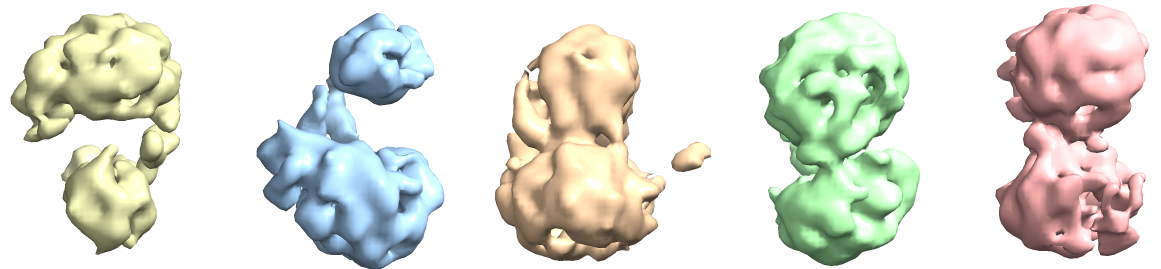
The maps are shown below:
_______________Homo Refine_________________________classe0 from 3rd Hetero Refine
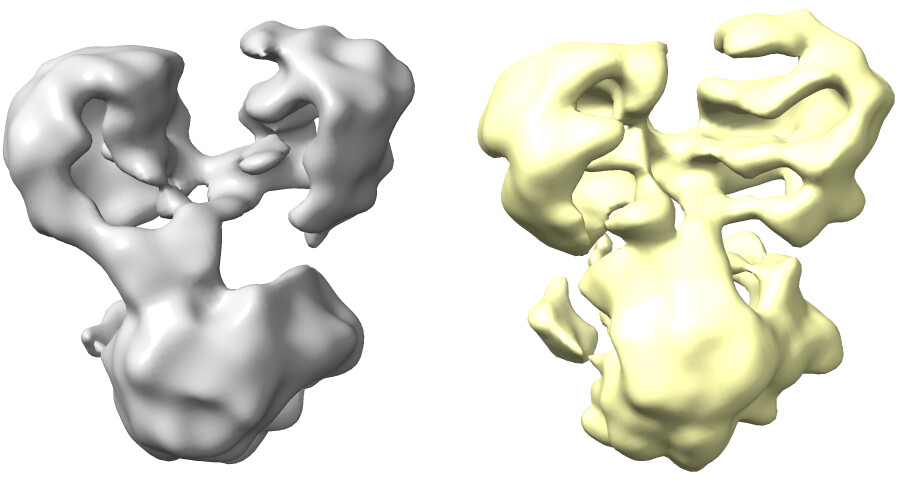
Is there anything I could try to get a better 3D reconstruction?