Hi all,
I am looking for a protocol to clean particle stacks in 3D - using mutli-class ab-initio and heterogenous refinement. Briefly:
I have a heterogeneous sample (monomer (127 kda), dimer, tetramer, filament (2-5 mDa)) ranging from 100 A - 500 A (or more), with some globular shapes, some oval shapes, square shapes (ringish) and a pseudofilament. I was first trying to discriminate the different complexes by 2D classification, but it was recommended to use 3D classification. I thought this would be the best because the box size for the pseudofilament is vastly different then what is needed for the monomer.
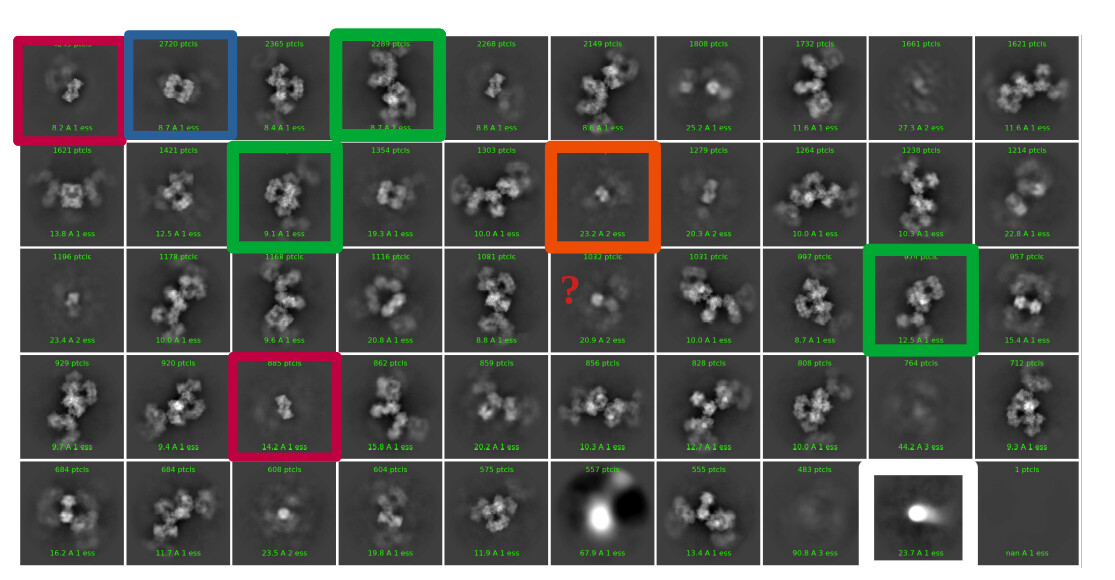
My thought is to select a picking protocol that gets everything, indiscriminately. Then, do many rounds of 2D classification with the largest box size required for the largest object (psuedofilament). Use 2D classification to get rid of bad picks, and anything that looks like a protein is kept. I would likely use 150 classes with default settings.
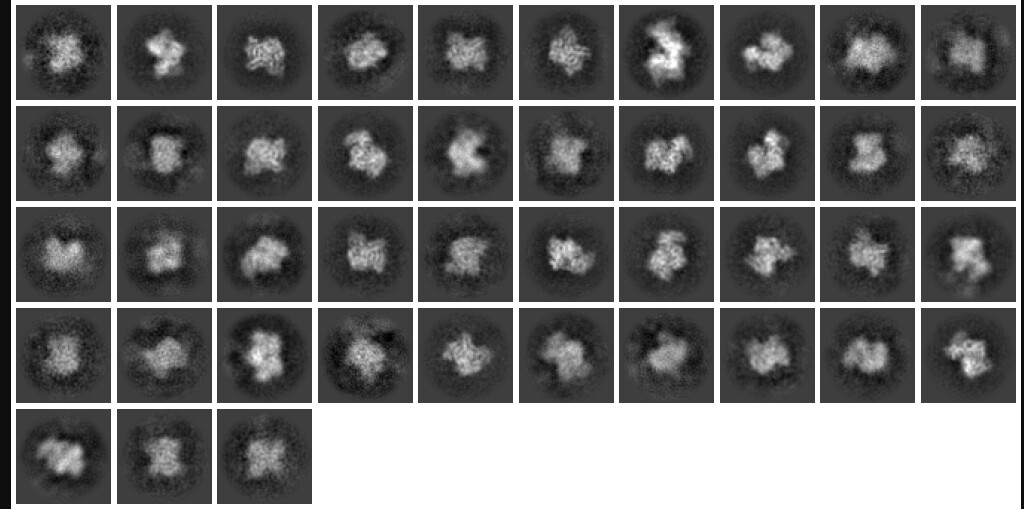
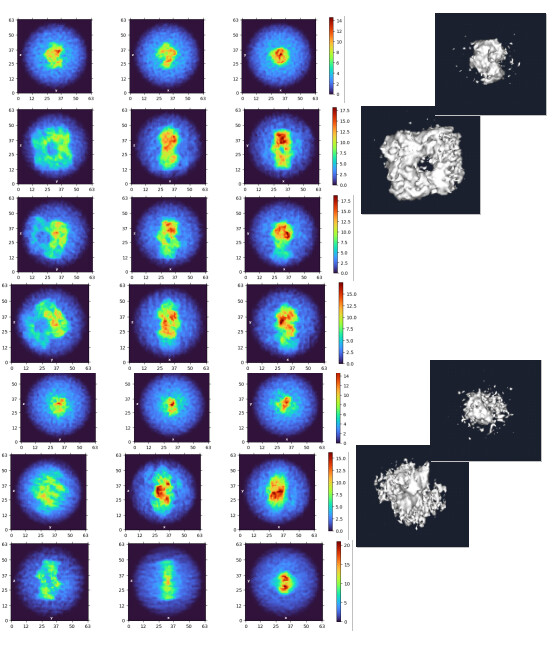
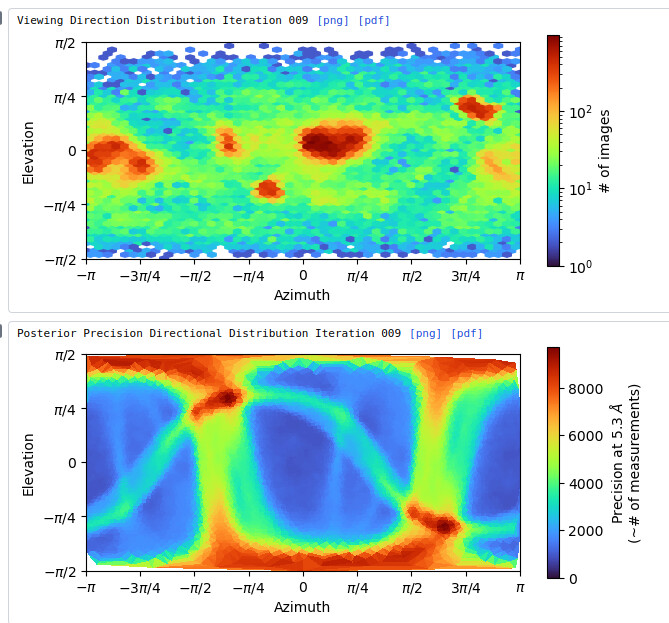
My question now is what sort of procedure/settings do I use for 3D classification? My thought is ab initio with 7 classes (3+ more than # of complexes), with class similarity = 0. Then use all particles and all volumes in heterogenous. Next, remove all bad refinements that look like noise? What metric/validation should be used to discard class/particles? I am not sure how to discriminate between poorly populated class and junk. Do you let multi-class ab initio give you a junk volume? I see commonly that it isn’t necessarily junk class, but rather splits a volume into two views. Then, redo this again…over and over until there are effectively no particles in the junk volume classes.
Is there something else I should be considering? Will the small particles (and their views) be selected with a large extraction box?
Matt