Hello all,
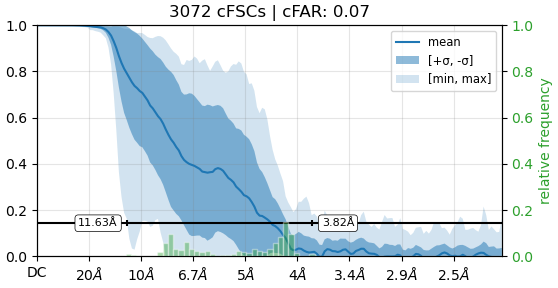
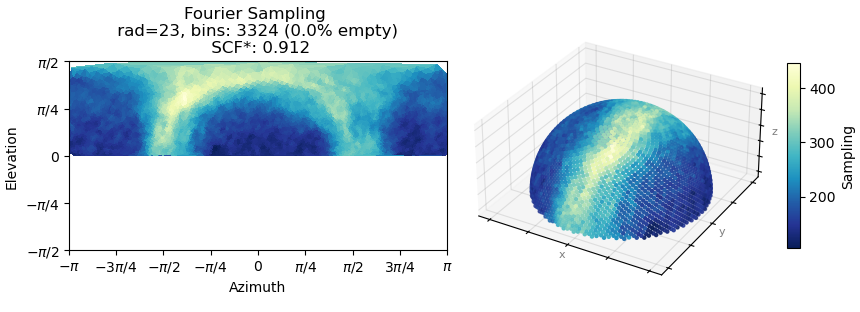
I am working on a receptor protein and have struggling with improvements to resolution to the map. The overall quality of the NU refinement is alright and the sharpened map that comes from this job looks great as I am able to fit a crystal structure inside. Looking at cFSCs, my cFAR score is very bad. There seems to be two populations of particles. When running orientation diagnostics, the SCF score is high (0.912), looking at the meaning of these values, this suggest that there is junk that is interfering with my refinements. I have tried heterogenous refinement to see if I can tease these out by running ab initio on these particles and then running heterogeneous to sort them out, but haven’t had good luck on it. Could I be approaching this from the wrong angle? Alternatively, I’m wondering if there is a way to sort out the particle list based on what the CTF values are? We are on version 4.6.0
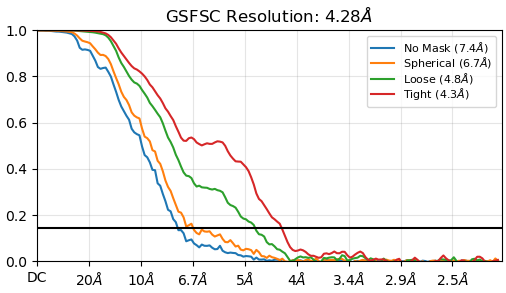
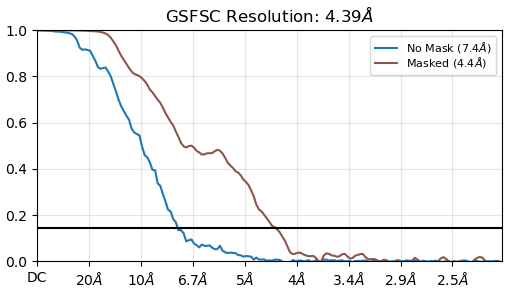
Below are my attached GSFSC from NU-refine and the cFAR from that same job.
If you need more info, let me know! Thanks all!
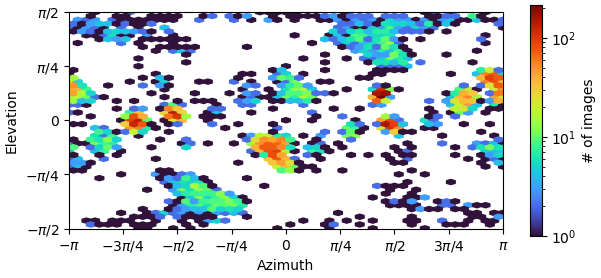
What does the orientation diagnostic look like? How many particles? If evenly dispersed, chances are there’s still lots of hidden junk, yes. Or your sample could be naturally heterogeneous and needs different conformations separating out. Could try 3D classification into a fairly high number of classes with a moderate resolution. If very few particles, could just be that there aren’t enough.
There are a few cases where reasonable/good maps have unusually poor cFSC values. This is know to the CryoSPARC developers and they’re investigating. 
The particle count is quite low, 57.8K particles, it was at 70.1K before (did some attempts at curation) to clean up.
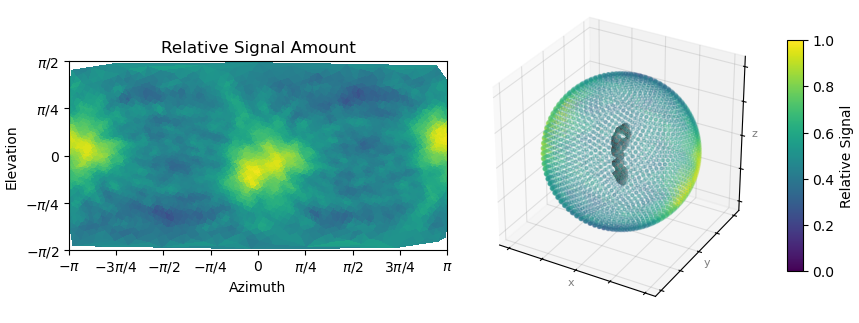
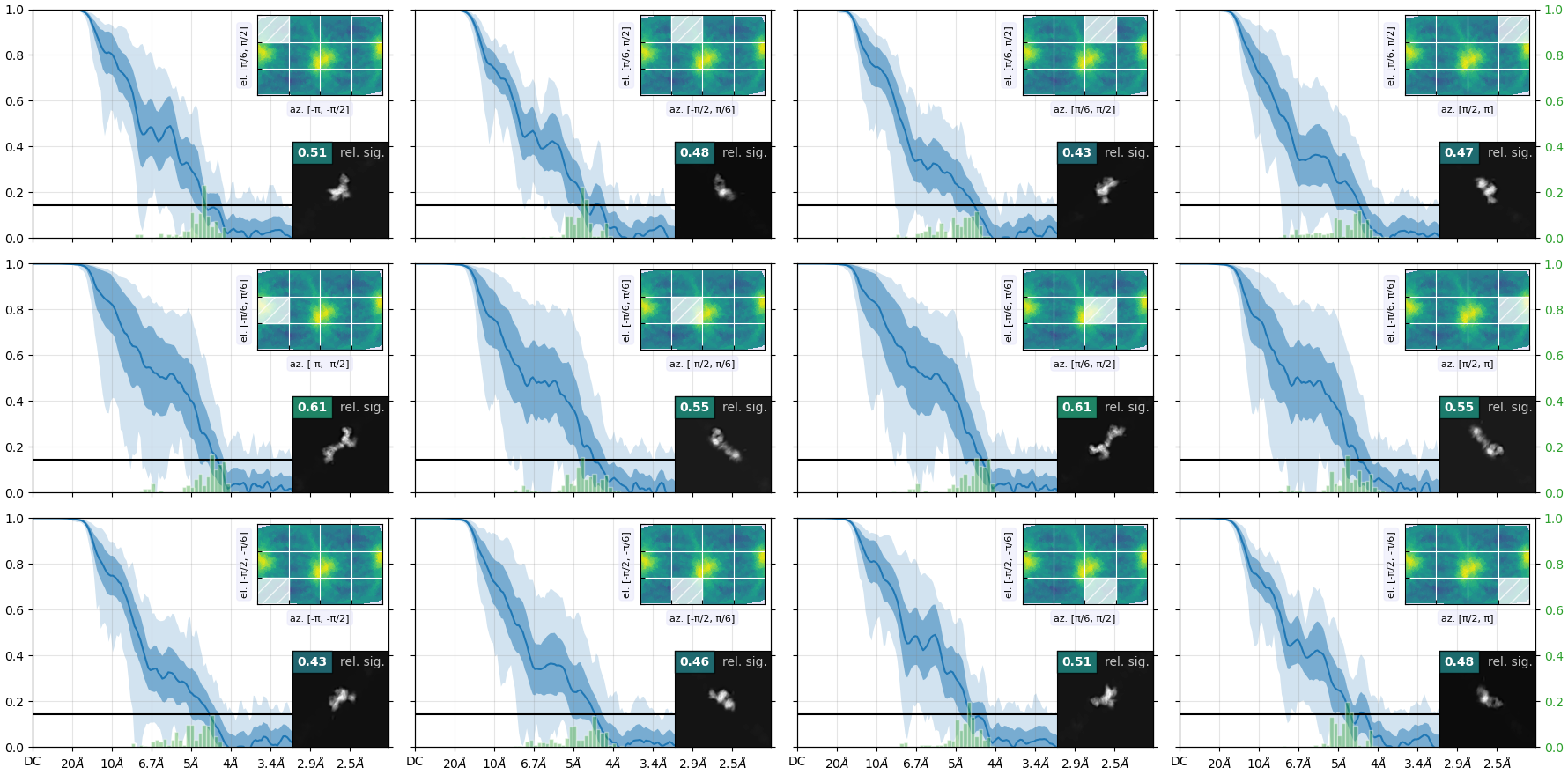
Here is the orientation diagnostics (showing what I think is important to take away from this job):
I have been learning towards simply just collecting more data. like you mention this could just be a particle count issue.
Sorry, I find the refinement plot easier to visualise (this one):
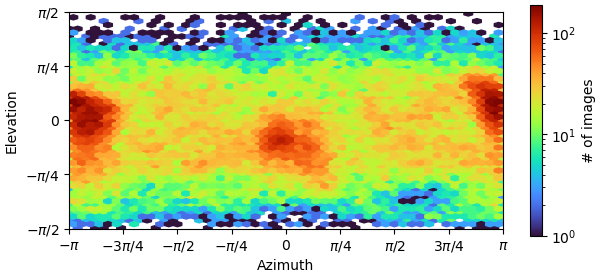
But from the plots you posted, if I’m reading them right, it looks like you’ve got a fairly strong preferred orientation.
Oop. Here is the orientation plot. Yeah, I do get a lot of side views. Likely because it is easier for me to validate that this is indeed my particle. The receptor is quite “skinny” and therefore top-down or down-top views are hard to distinguish with high confidence during 2D upstream.
Yes, if you folded that round, you’d find you’ve got a fairly strong preferred orientation. You could try Rebalance Orientations to reduce them somewhat, and re-refine (perhaps local refinement…?). Lots of samples demonstrate avoidance/disfavouring of certain orientations but still refine to a reliable, high resolution map (photosystems are a good example) because the “equatorial” (side) views are well dispersed around the spherical belt. “Polar” (top) views are still desirable but are not critical. The issue for you with the cFSCs is related to (a) that strong preferred orientation at pi/-pi and (b) lower particle count.
1 Like
There are some tricks that I may try on my next grids that our lab has worked out with preferred orientation with other receptors. Good suggestions to try. Thanks for the insights! Much appreciated
Good luck with the biochemical optimisation! That’s usually the best way to solve it.