Oh great, didn’t know that one can select the “Building” badge.
Some limited succes, now it failed at the next “Extract from micrographs” job.

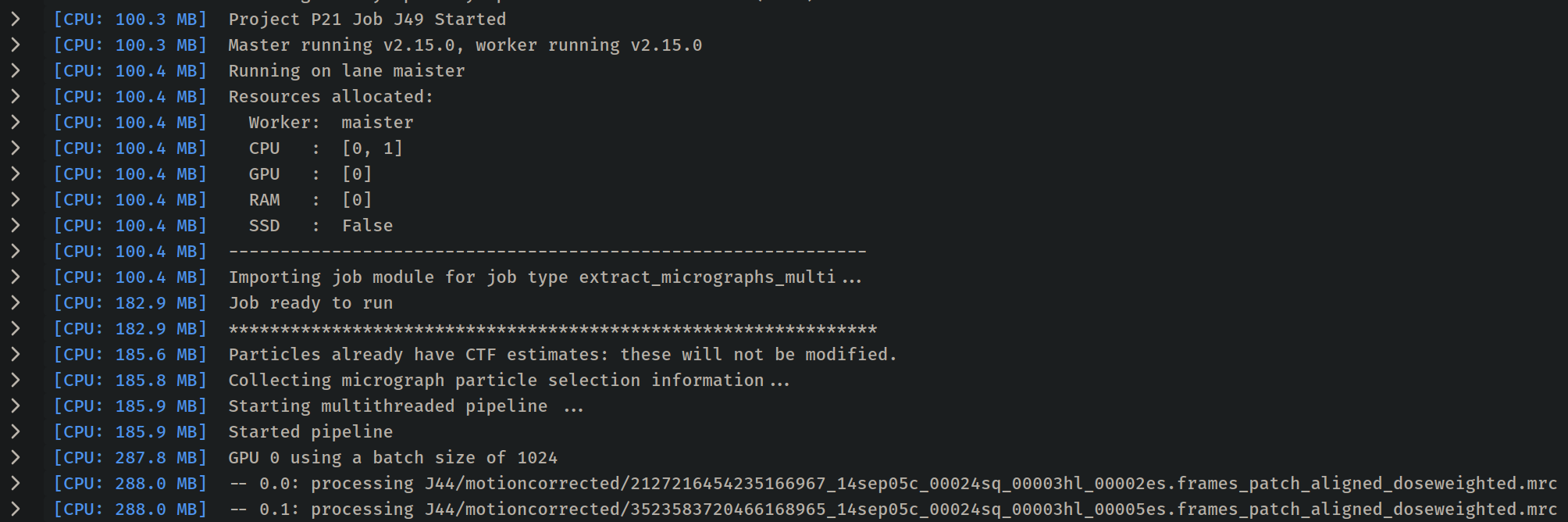
A bit more descriptive this time:
[CPU: 184.6 MB] Starting multithreaded pipeline ...
[CPU: 184.8 MB] Started pipeline
[CPU: 285.4 MB] GPU 0 using a batch size of 1024
[CPU: 285.6 MB] -- 0.0: processing J61/motioncorrected/13054666239615727002_14sep05c_00024sq_00003hl_00002es.frames_patch_aligned_doseweighted.mrc
Writing to /cs_extensive_workflow/P21/J71/extract/13054666239615727002_14sep05c_00024sq_00003hl_00002es.frames_patch_aligned_doseweighted_particles.mrc
[CPU: 285.6 MB] -- 0.1: processing J61/motioncorrected/16675970042098428134_14sep05c_00024sq_00003hl_00005es.frames_patch_aligned_doseweighted.mrc
[CPU: 876.6 MB] -- 0.0: processing J61/motioncorrected/2465388814133724455_14sep05c_00024sq_00004hl_00002es.frames_patch_aligned_doseweighted.mrc
[CPU: 877.2 MB] Traceback (most recent call last):
File "cryosparc2_compute/jobs/runcommon.py", line 1685, in run_with_except_hook
run_old(*args, **kw)
File "/opt/cryosparc2_worker/deps/anaconda/lib/python2.7/threading.py", line 754, in run
self.__target(*self.__args, **self.__kwargs)
File "cryosparc2_compute/jobs/pipeline.py", line 65, in stage_target
work = processor.process(item)
File "cryosparc2_compute/jobs/extract/run.py", line 238, in process
_, mrcdata = mrc.read_mrc(path_abs, return_psize=True)
File "cryosparc2_compute/blobio/mrc.py", line 135, in read_mrc
data = read_mrc_data(file_obj, header, start_page, end_page, out)
File "cryosparc2_compute/blobio/mrc.py", line 98, in read_mrc_data
data = n.fromfile(file_obj, dtype=dtype, count= num_pages * ny * nx).reshape(num_pages, ny, nx)
MemoryError
[CPU: 877.2 MB] Traceback (most recent call last):
File "cryosparc2_compute/jobs/runcommon.py", line 1685, in run_with_except_hook
run_old(*args, **kw)
File "/opt/cryosparc2_worker/deps/anaconda/lib/python2.7/threading.py", line 754, in run
self.__target(*self.__args, **self.__kwargs)
File "cryosparc2_compute/jobs/pipeline.py", line 65, in stage_target
work = processor.process(item)
File "cryosparc2_compute/jobs/extract/run.py", line 271, in process
cuda_dev = self.cuda_dev, ET=self.ET, timer=timer, batch_size = self.batch_size)
File "cryosparc2_compute/jobs/extract/extraction_gpu.py", line 176, in do_extract_particles_single_mic_gpu
output_g[batch_start:batch_end] = ET.output_gpu.get()[:curr_batch_size]
File "/opt/cryosparc2_worker/deps/anaconda/lib/python2.7/site-packages/pycuda/gpuarray.py", line 287, in get
ary = np.empty(self.shape, self.dtype)
MemoryError
[CPU: 1.06 GB] (1 of 20) Finished processing micrograph 0.
Do you need any additional logs?
Thank you a lot!