I am working on a homodimeric protein, each monomer ~75kDa (total size 150 kDa).
We have conducted a high number of biochemical and biophysical analyses of this protein, and it is well-behaved. Comes out of the mass photometer, SEC-MALS, etc., as a nice peak, dimeric. It’s active and responds as expected in assays etc. Point being - it’s not an unfolded mess of protein.
However, solving its structure is proving problematic.
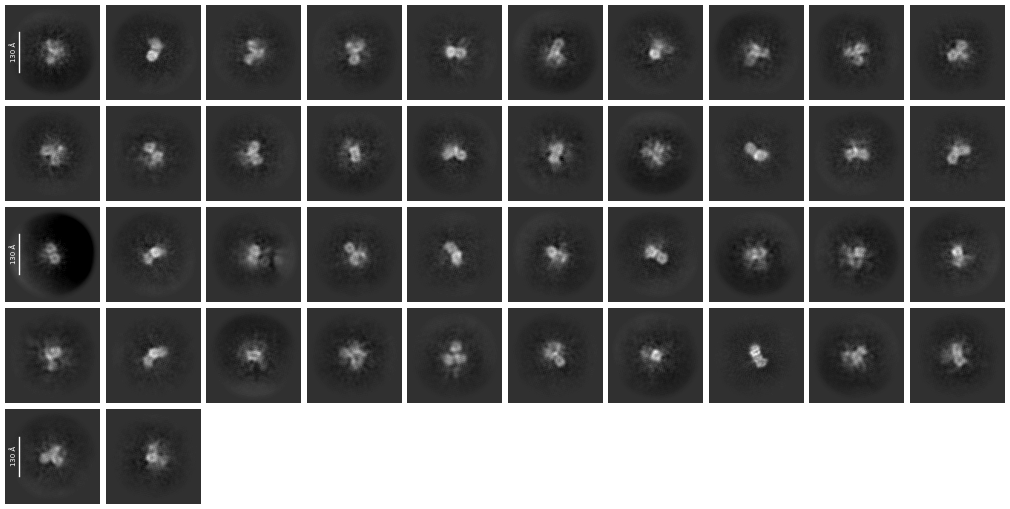
We keep getting “hollow” structures, often with very few structural details. From the 2D Classes, we frequently have three three-lobe structures, which are very much in line with the predicted structure. Ab initio structures overlayed with the alphafold structure produce what you would expect - a three-lobed but “hollow” structure - each of the lobes has massive holes going through them.The 2D classes also appear to show this also, there are block dots in the middle of each lobe.
We have conducted numerous datasets now, and achieved very similar results. Starting with ~2M particles, we whittle it down to ~150k and produce very poor results.
Would anyone have any advice? Are the poor results due to inherent flexibility in the protein? Should we cross-link to reduce this? Is this possibly a phase issue, or something to do with the potential 2D symmetry of the dimer? Any advice would be much appreciated.
From my own experience this seems unfortunately like noise. I had a similar situation with a very well behaved protein but I could never see anything resembling intact particles on a micrograph, no matter the vitrification technique. When I would refine my “best” volumes (which were never very convincing) I would just end up with hollow shells full of holes. The protein behaved very well in solution but could not handle vitrification at all.
I guess some questions beforehand:
Can you clearly see your particles in the micrographs? Are they truly convincing?
Do you see any aggregation, or perhaps sandy clouds on the micrograph?
It doesn’t appear to be the case, but is your sample membrane bound? If it is, have you tried differing detergents?
Were your particle picks unbiased or did you use some template? Template picks can yield convincing low resolution structures, but if the actual particles don’t match then the higher resolution detail will be missing.
I do see particles, and we don’t get this structure on an empty grid. We have also added compounds/protein activators/inhibitors and get variations on this structure which make sense too. I don’t think it is noise but it’s not far from my thoughts.
I would pick some of your best 2D classes for a template picking, followed by extraction and 2D classification. Probably you want an “extensive” 2D classification instead of using the default parameters (e.g. number of classes 200, batch per class 400, number of on-line iterations 40).
My experience is that you probably need to see clear SS features in your 2D classes to get decent ab initios.