I have found previous posts to be decently helpful in analyzing my SPA dataset (a 150 kDa protein complex with expected C1 “symmetry”). The protein has 3 domains which I’ve confirmed with SEC-MALS, SDS-PAGE, and other methods. At first, I found my results promising after I did a manual pick and 2D classification. I get 3 domains as expected. (2 of the domains are obligately bound, while the 3rd might come off.) However, when I use my manual picks and train a Topaz model for automated particle picking, I start to lose 1 of the domains. This could be possible, since 1 of the domains is reversibly bound. However, I’m wondering if this might be caused by some artifact of overfitted particle picking and/or alignment issues. I’m attaching some images of the workflow below.
First round of manual picking:

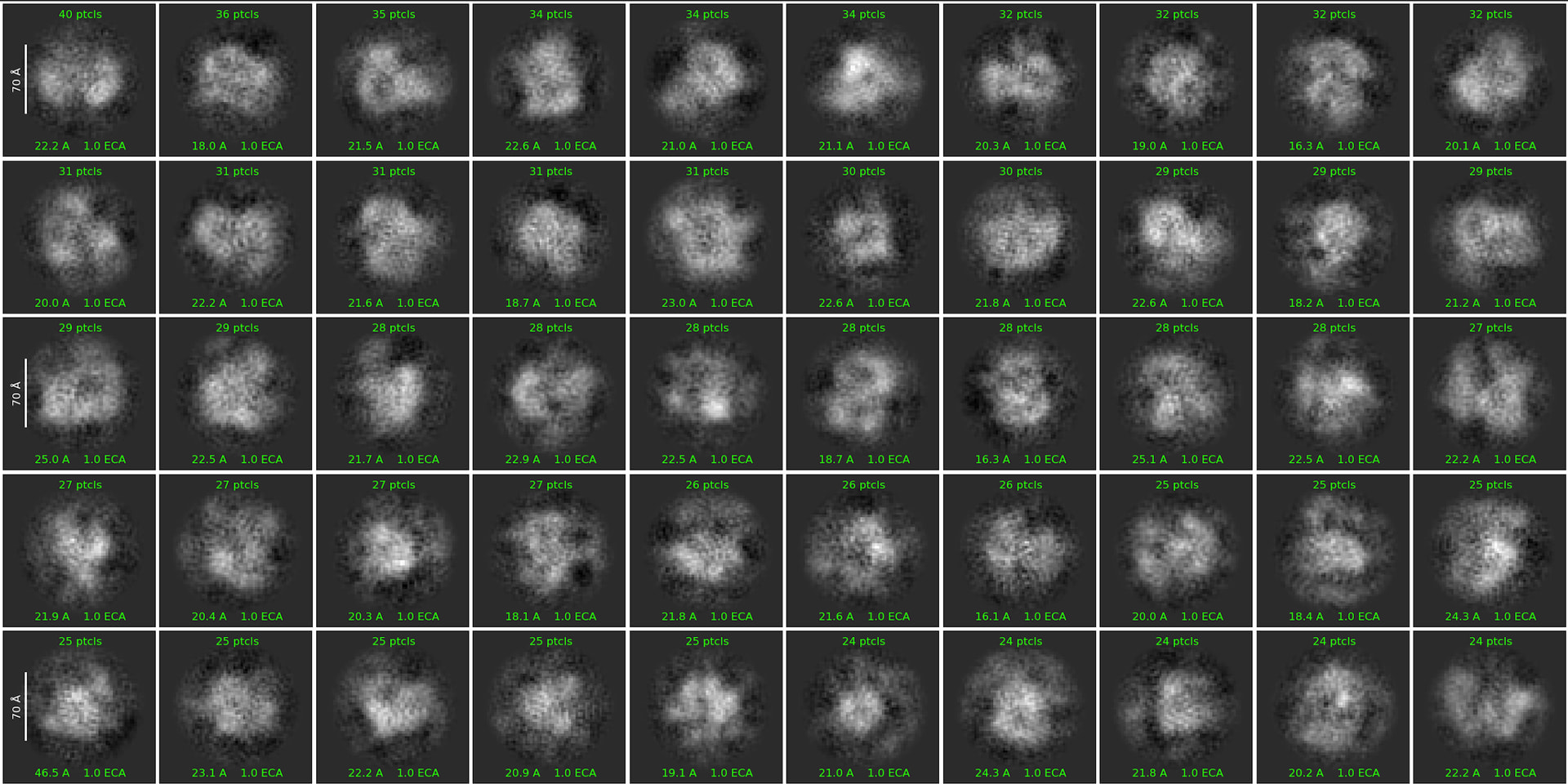
First round of 2D classification with purely manually picked particles which showed promising 3-domain classes (2,344):
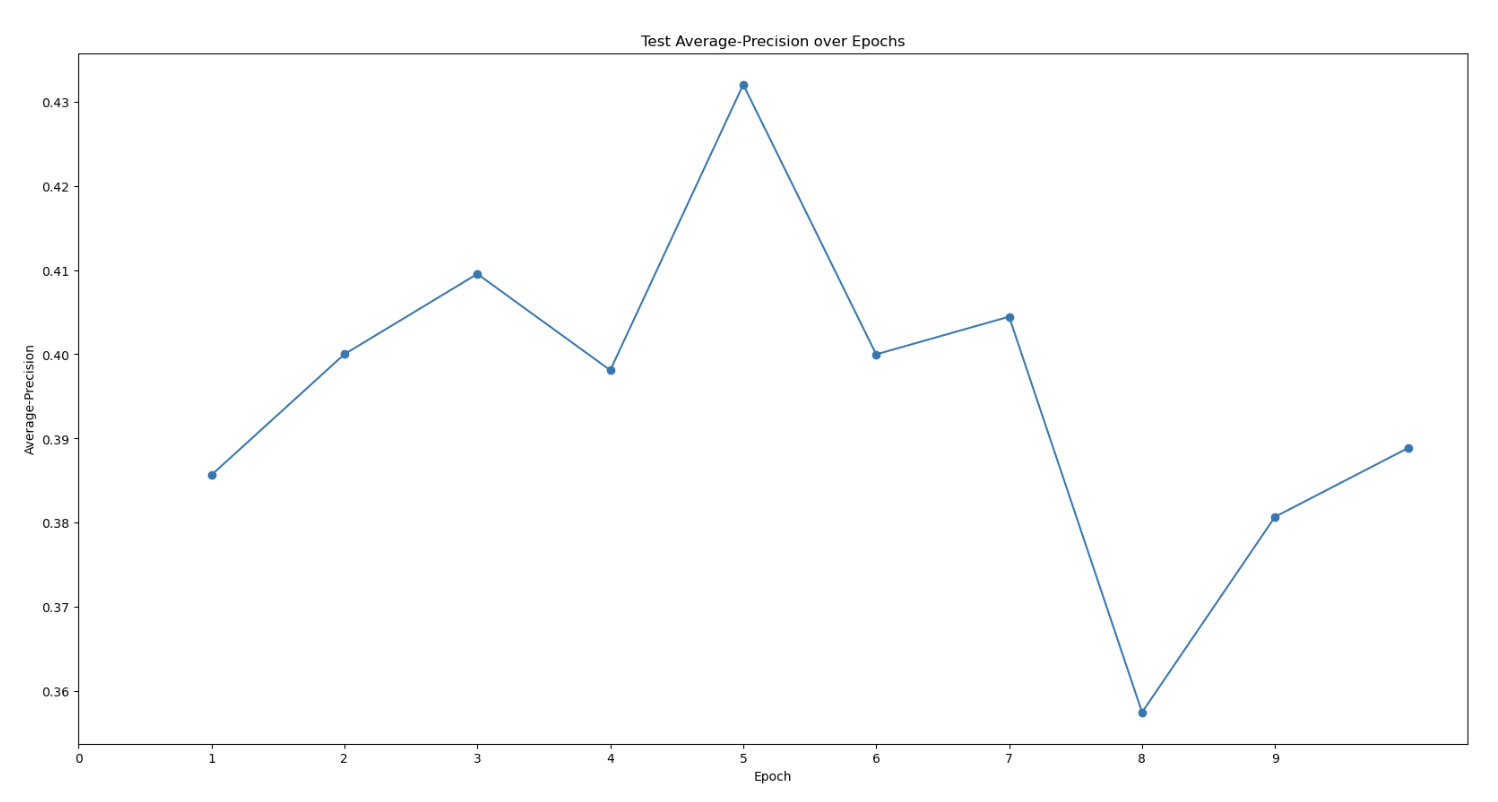
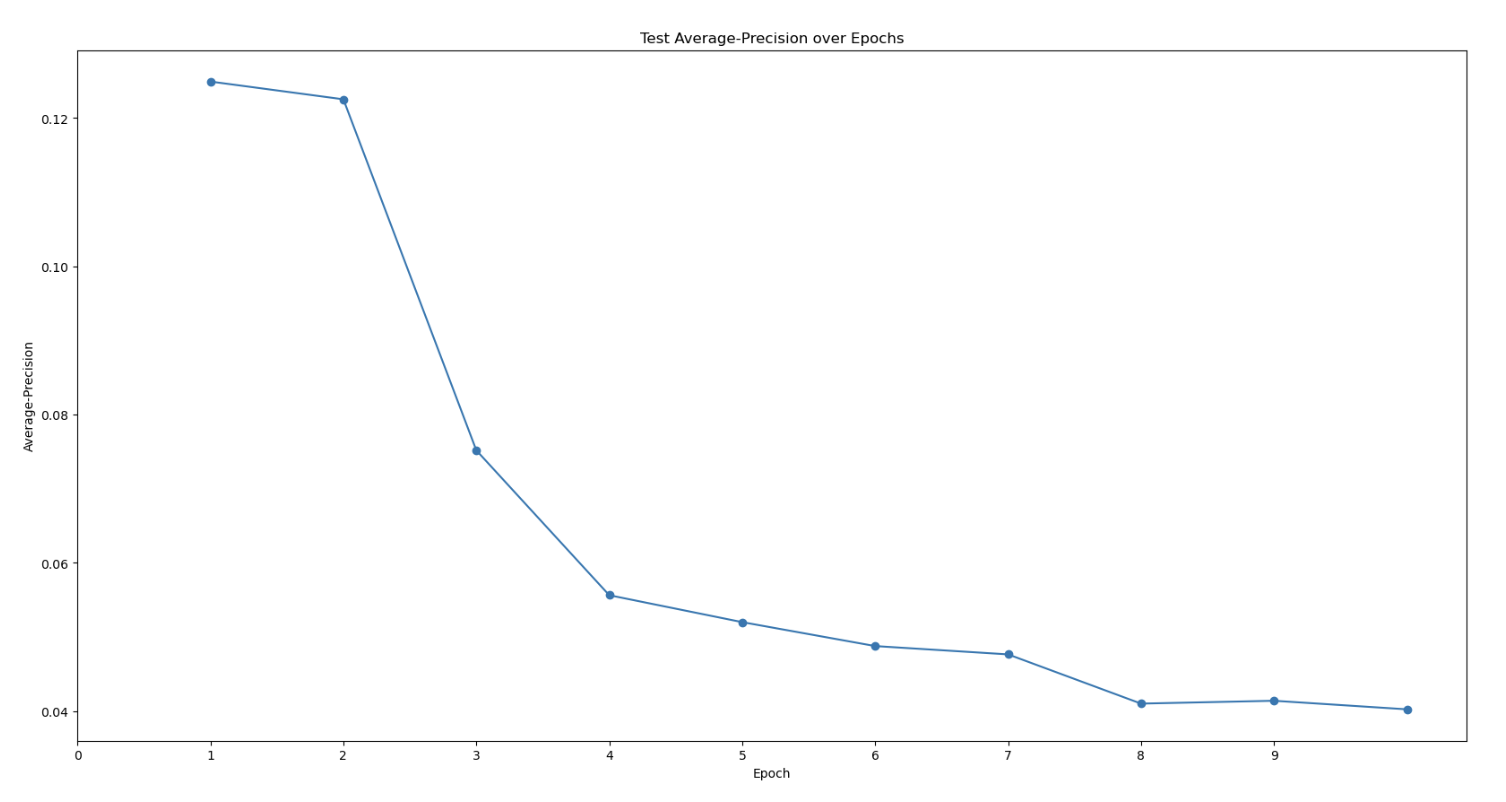
First topaz model’s predictive performance:
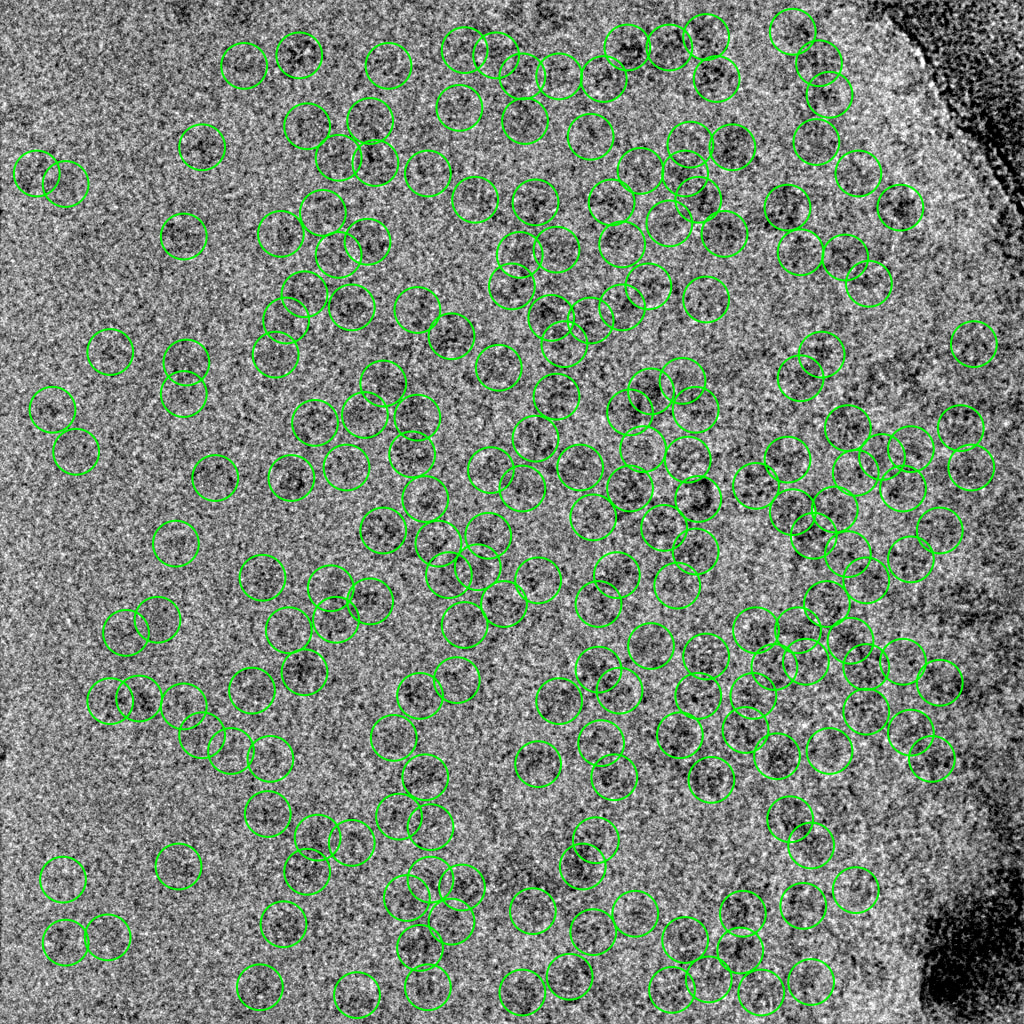
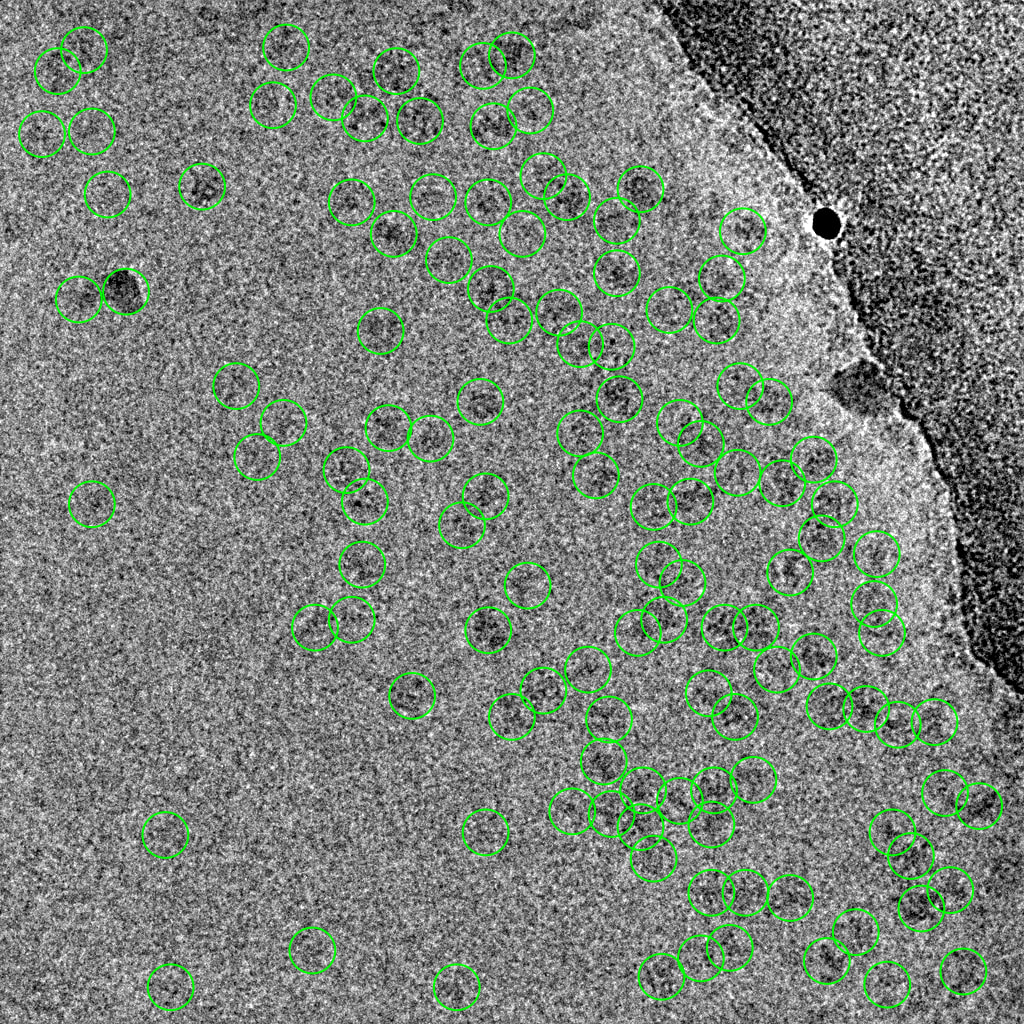
2 representative micrographs that show this Topaz picking model’s strategy:

First round of 2D classification with automated particle picks (via Topaz):
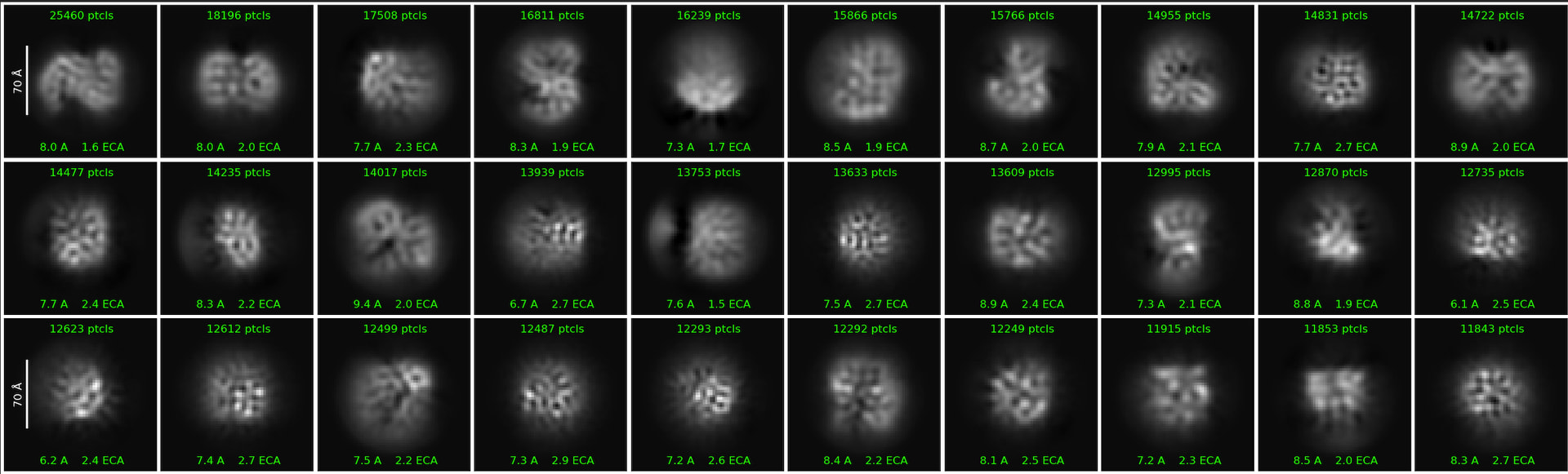
At this point, it looked somewhat promising, though not as good as manual picking. However, I start encountering issues when I select some 2D classes (from the above) to then re-train a new Topaz model. I can’t seem to get a better Topaz model.
Additionally, according to my 3D volumes in ab initio reconstruction, I might not have too much of the 3rd domain. Out of 3 initial volumes, roughly 1/3 of the particles have this 3rd domain. (This is also concerning in its own right, since I would have expected there to be some skewing of the distribution.)
Are there any strategies to increase the Topaz model’s particle-picking accuracy and workflows to increase the detection of that 3rd domain? My models’ max at ~0.4 AUC-PR seems to be on the lower end. I am also wondering if there’s some weird alignment issue because one of my protein’s domains. My hypothesis is that the 2 obligately bound domains have a very distinct “backwards S” shape that allow it to be easily classified in 2D classification. However, the full trimeric complex might look more globular and lose this “backwards S” shape, meaning that noise can easily be classified into the same 2D classes that correspond to the full trimeric complex. I’m trying to be unbiased when I look at my micrographs. I just see so many 3-domain particles to think that the full complex is only a minor species… (As you can see in the manually-picked particles that were 2D classed, the densities are weak with only ~2k particles, but you can clearly see there’s a bias toward seeing all 3 domains…)
So far, the thing that has helped the most is to set “force max poses” off. I appreciate those who have suggested that in previous posts — thank you!
