Hi, community.
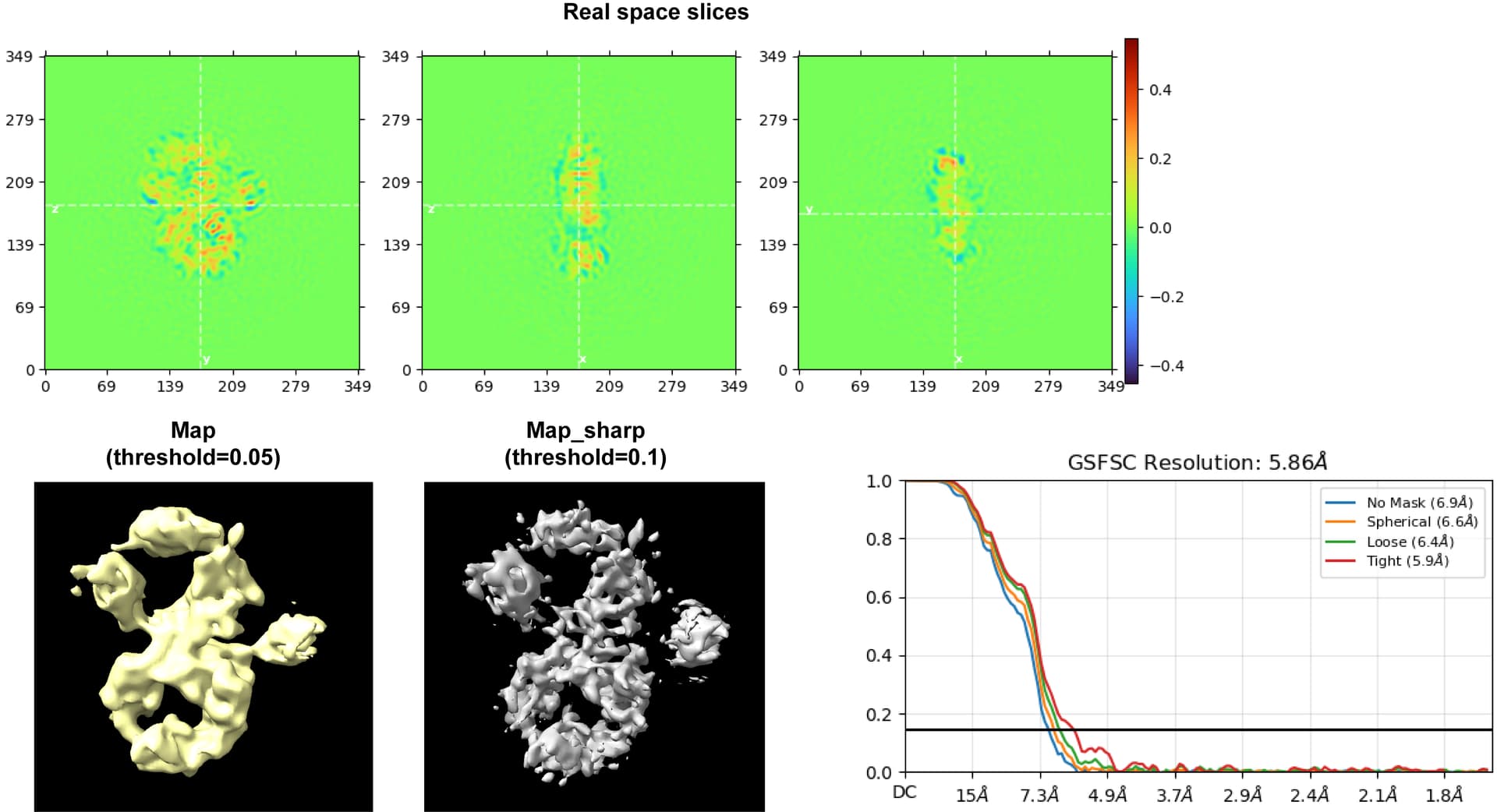
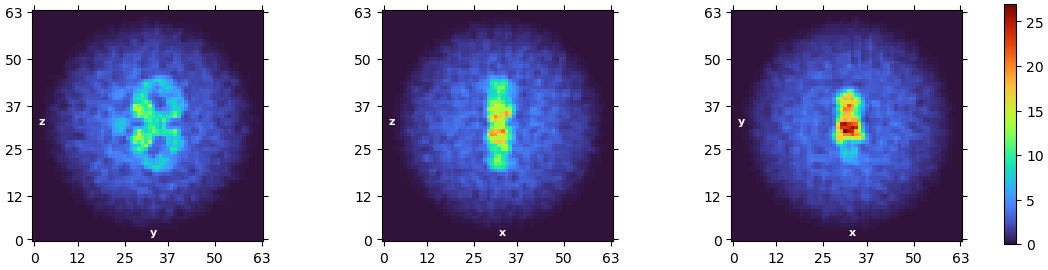
I am currently working with a small (150kDa-ish) protein, and I got decent 2D classes, OK initial models, but poor models after all kinds of refinement. Specifically, the refined map is always either incomplete or chunky and lack of details, which doesn’t resemble the real space slices during processing or resolution determined from the GSFSC curve. While I just discovered that the map_sharp is much clearer. Pls see an example below:
The same issue happened to all my refinement tasks and even the 3D flex tasks. In brief, the model generated by 3D flex was chunky as it was, despite a 3.73A resolution estimation.
I am wondering why the refined map is so crude and doesn’t match the real space slice (you can see the peptide-like patterns from the slice, which cannot be seen at all on the map. And also, why would the map and map_sharp make such a difference.
I am willing to provide additional information if needed. Thanks for any help in advance.
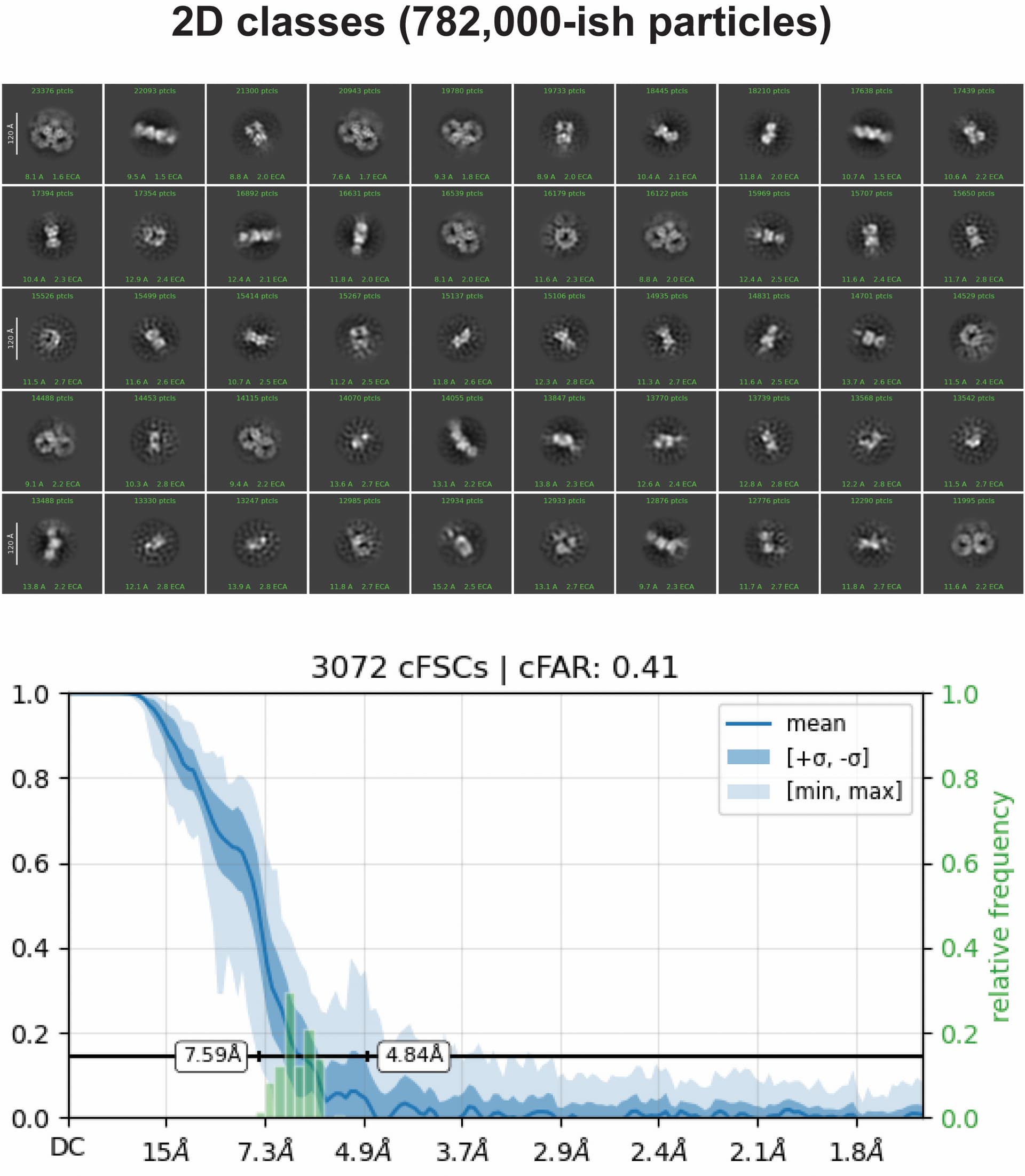
The 2D classes would be nice to see, as I don’t think that sharpened map is particularly trustworthy. What do the cFSCs look like? The real space slices look very anisotropic. The left and right “wings” of the 3D also look very overfitted, with the rest of the map a little less so.
What is the predicted secondary structure? Mostly beta sheet or are there some helices in there?
There would just be a couple of alpha helices (On the upper part of these two wings, close to the cap), and the majority of the protein (I would say, >75%) is composed of beta strands/ sheets. The strange density blob on the right is probably a very flexible linker flipping around.
Those 2D classes are still extremely heterogeneous. You might find using a large number of ab initio classes, followed by a long heterogeneous refinement teases out the different states/mixes.
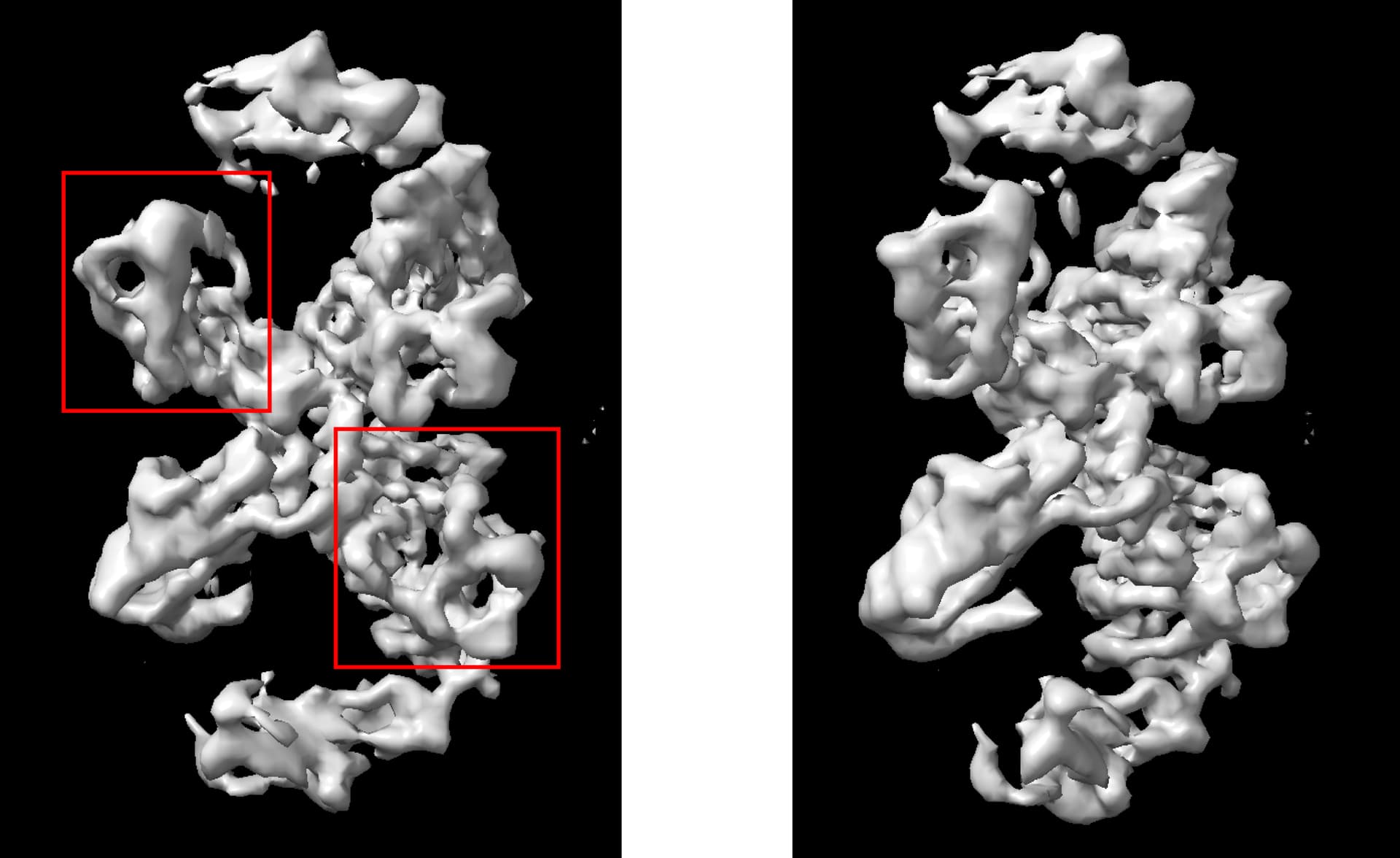
Sorry for the late reply. I tried out your suggestions by running 5 initial models, picking the best one, and subsequently running NUref. The resolution does seem better. Thank you so much for your help! Pls see attached:
However, I ran into another trouble that during refinement, the domains (highlighted with red boxes) are swapped. I was able to get it fixed by generating symmetrical (C2) initial models and running symmetrical refinements. But the resolution will drop, and b factors can get crazy (>300) in that case. I also tried symmetry relaxation function, but the misalignment issue would recur.
When you say the domains are swapped, what do you mean exactly? Is it possible that you started from an ab initio model with a different (inverted) hand? In that case you can just flip the volume using volume tools and redo refinement
The molecule is C2 symmetrical (or nearly symmetrical) along the vertical axis, in which the larger domains should be upper, and the smaller ones should be lower. Due to some reasons, now the smaller domains are assigned on the left, while the larger domains are assigned on the right.
I found a similar issue also happened to my initial model actually. I was able to “force” the assignment by setting a C2 symmetry (pls see attached)
I tried flipping the initial model, but the result was unchanged. To be honest, I just feel that there are some misalignment issues with both initial modeling and subsequent refinements.
I still feel from the 2D that it’s much more heterogeneous than a single structure… I wouldn’t be worrying excessively about handedness or “swapping” just yet…
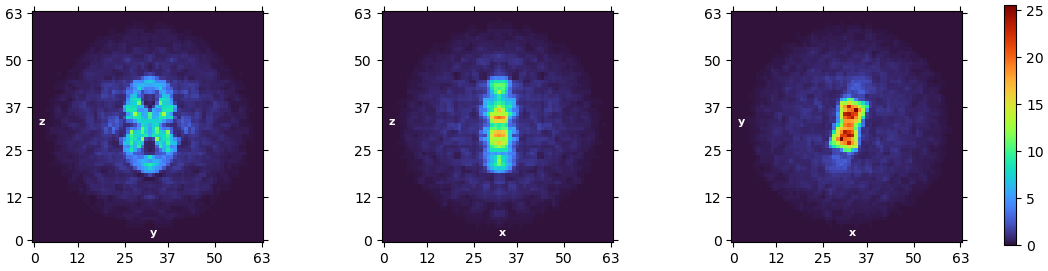
To clarify, I established 5 initial models out of the particle stack and ended up only refining the best model with the according particle sub-stack. By doing that, I was able to get rid of bad classes that look like junk or incomplete proteins. I didn’t really run 2D classification with that subset, but it must be much purer than what I showed above. (The initial model is shown below).
Pls also see the 3D model I attached above. The 2nd structures in the individual domains are visible now, and they match resolved structures of similar type. But due to whatever reasons, the arrangement of domains were problematic (pls see my previous post, in brief, the molecule is nearly C2 symmetrical along the long axis, which is mis-assigned in my models). Actually, I found the issue started to pop up in the initial model as well. by engaging a C2 symmetry in model building and refinement, I was able to enforce the right symmetry, but the resolution and b factors are bad.
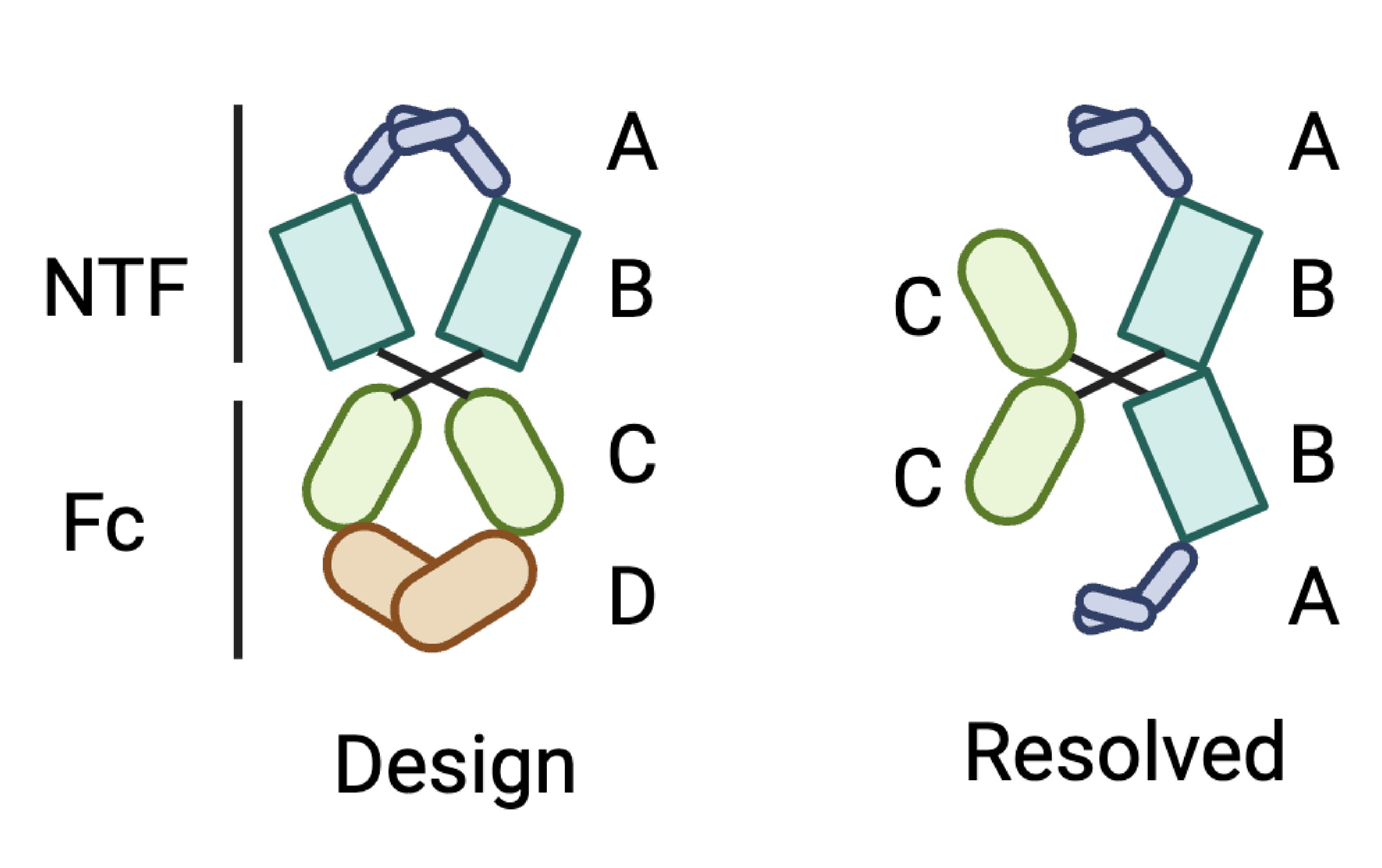
It doesn’t seem like a good practice to ask about other technical issues after the thread is resolved… But just to make it clearer, the issue can be summarized by the following picture:
Basically, the domain D is gone in resolved structure. And both top and bottom now become partial domain A. It totally makes no sense. Since first, the Fc is very stable, and domain D contains His6 tag, by which we purify the protein. And the domain C is just too small to form a complete ring with domain A and B.
If you don’t have any ideas at this point, I will start a new thread.
Hi Tingzhen, this helps explain it, thanks. I agree it is puzzling, but I would avoid enforcing C2 when it is apparent from the initial volume that it is not C2 - especially as resolution worsens after enforcing C2.
i agree with @rbs_sci that there is unresolved heterogeneity- not just junk, but different structured species, which if resolved may help you solve the puzzle. E.g. in your initial 2D, the first class seems to be a higher oligomer of some sort, while some of the later species seem to be broken or incomplete particles.
Performing ab initio with subsets of classes that visually correspond to separate species, followed by heterogeneous refinement, may be helpful.
Yes, Oli. After I got back and took a closer look at my 2D averages, I found there were several classes that do look like my final model, meaning the misalignment issue happened earlier than I expected… I probably need to get back and repeat the 2D classification to fix these bastards… They not only duped the computer, but also me… Do you have any suggestions in aligning such small particles (120A in diameter)?
BTW, here are my parameters:
50 classes, circular mask diameter 150A, force max over poses/shifts OFF, 40 online EM iterations, batch size per class=400, all the rest parameters unchanged.
I would go back one step further and take a look at your picking - how did you pick? On denoised mics or regular? That is the most likely origin of any systematic miscentering IMO
I generated templates with the a model (with low resolution) I built previously. Then I applied template picker on regular movies. It seems that the trouble might be caused by the shitty templates. I am planning on generate a good model and use that to generate new templates. Any other suggestions?
If you haven’t already, I would try denoising your mics using the Micrograph Denoiser, then template picking on the denoised mics. It can make a significant difference to the quality of the resulting picks.